
BioInformatics:中科院生化细胞所陈洛南研究组建立自适应整合多组学数据进行样本模式预测的新
2017-04-18 佚名 生物帮

2017年3月28日,国际学术期刊《BioInformatics》在线发表了中国科学院生物化学与细胞生物学研究所陈洛南研究组题为“Pattern fusion analysis by adaptive alignment of multiple heterogeneous omics data”的最新研究成果。该成果展示了基于流形思想,通过整合多种组学数据实现自动的信息融合与偏差校正,并从中识别出
2017年3月28日,国际学术期刊《BioInformatics》在线发表了中国科学院生物化学与细胞生物学研究所陈洛南研究组题为“Pattern fusion analysis by adaptive alignment of multiple heterogeneous omics data”的最新研究成果。该成果展示了基于流形思想,通过整合多种组学数据实现自动的信息融合与偏差校正,并从中识别出全局样本模式的方法。
随着测序技术的发展,组学数据呈现井喷式增长,大数据时代的来临给生物信息科学带来了新的机遇和挑战。传统的基于单组学数据的研究,只能够有限地解释生物系统或者复杂疾病的特征;采用的数据类型不同,也往往得到不同的分析结论;这些均大大阻碍了高通量技术在临床方面的应用。既然各组学数据对于刻画生物表型的贡献不尽相同,那么,能够将不同类型的组学数据有效地整合到一起,以综合和整体的视角来看待生物过程或者表型,成为了信息转化医学应用的一个重要突破口。尽管,近几年来有一些研究成果问世,但这些成果大都基于特定的模型假设,在整合不同信噪比的组学数据时,往往容易遗漏重要信息,甚至得到错误结果。
针对整合组学数据中存在的此种难题,陈洛南研究组开发了一套新的整合办法。该方法从数据本身出发,在保留各组学的生物特性的前提下,将各层数据反映出的样本模式进行自动非线性整合,并且由优化得到符合数据固有特征的全局样本模式。其中,各组学数据对于整合结果的贡献得以量化,数据偏差得以校正,可以有效地鉴别出数据内部结构特征。这种无特殊假设的计算模型,可真正实现数据驱动的整合。研究组不仅在数字实验上验证了该方法的有效性,还成功应用到生物学数据的分析——癌症细胞系CCLE数据与临床病人TCGA数据上,揭示出整合后的癌症分型具有更显着的生物学或者临床预后的重要性。
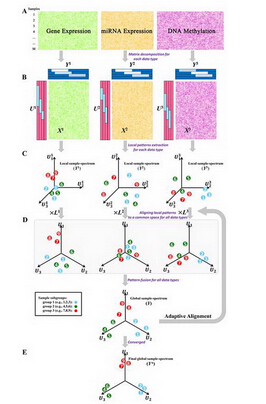
自适应整合多组学数据进行样本模式预测
原始出处:
Shi Q, Zhang C, Peng M,et al.Pattern fusion analysis by adaptive alignment of multiple heterogeneous omics data.Bioinformatics. 2017 Mar 28.
小提示:本篇资讯需要登录阅读,点击跳转登录
版权声明:
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言





#Bio#
88
#样本#
98
#自适应#
106
#中科院#
74