
腓骨肌萎缩症:症状与体征、病因、流行病学、诊断与治疗
2022-09-15 MedSci原创 MedSci原创

腓骨肌萎缩症(Charcot-Marie-Tooth disease,CMT) 是一组遗传性周围神经病。目前已发现的致病基因达 60 余种。其主要特点为慢性进行性、长度依赖的运动及感觉神经病, 最常见
腓骨肌萎缩症(Charcot-Marie-Tooth disease,CMT) 是一组遗传性周围神经病。目前已发现的致病基因达 60 余种。其主要特点为慢性进行性、长度依赖的运动及感觉神经病, 最常见表现为下肢起病的、缓慢进展的肢体远端肌肉萎缩,无力和感觉缺失。根据上肢运动神经传导速度主要分为髓鞘型和轴索型。根据遗传方式、临床表现以及电生理,CMT 主要亚型包括 CMT1-4 以及 CMTX。此外还有 CMT5-7、dHMN(远端型遗传性运动神经病)、HNPP(遗传压迫易感周围神经病)。在每个亚型中,不同字母代表不同基因突变(如CMT1A,CMT1B)。
一、一般概述
Charcot-Marie-Tooth(CMT)疾病是一组疾病,其中运动和/或感觉周围神经受到影响,导致肌肉无力和萎缩,以及感觉丧失。症状首先发生在腿部远端,后来发生在手部。由于神经轴突的异常或轴突周围的绝缘层(髓鞘)的异常,患有这种疾病的人的神经细胞不能正常发送电信号。在CMT中,特定的基因突变是导致周围神经功能异常的原因。在许多形式的CMT中,这些基因是已知的,而在其他形式的CMT中,虽然该病已知是遗传的,但具体的基因还没有被确定。
二、症状与体征
CMT疾病的症状通常在青春期逐渐开始,但也可以更早或更晚开始。在几乎所有的病人中,最长的神经纤维会首先受到影响。随着时间的推移,受影响的人可能会失去对脚、手、腿和胳膊的正常使用。常见的早期症状和体征包括对热、触觉或疼痛的敏感性下降,手、脚或小腿肌肉无力,精细运动技能出现问题,高步态(足下垂),小腿肌肉质量下降,经常绊倒或跌倒,锤状趾,高足弓和扁平足。伸展反射可能丧失。这种疾病是缓慢进行的,而且病情多变,受影响的人可能多年来一直保持活跃,寿命正常。在最严重的情况下,呼吸困难会加速死亡。
三、病因
众所周知,CMT是一种由影响其功能的异常变化的基因引起的遗传性疾病。现在已知有100多个基因对各种形式的CMT负责。一个单一的基因,即PMP22,在重复时,是大约50%的CMT病例的原因,而导致CMT的一些基因的破坏性变化可能极其罕见,只在少数家庭中发现。在大约40%的CMT病例中,还没有发现负责任的基因。
遗传性疾病是由父亲和母亲的染色体上的单一基因或某一特定性状的基因组合的变化引起。目前,只有在单一基因导致CMT的情况下才能确定其遗传原因,但在怀疑有多个基因发生破坏性变化的影响时,即所谓的多基因遗传,则不能确定。引起CMT的单一基因可以以常染色体显性、常染色体隐性、X-连锁或X-连锁显性模式遗传。
显性遗传病是指只有一个不工作的基因的单拷贝才能引起某种疾病。非工作基因可以从父母任何一方继承,也可以是受影响个体的基因改变(变异)的结果。每一次怀孕,受影响的父母将非工作基因传给后代的风险是50%。这种风险对男性和女性来说是一样的。
当一个人从父母双方各继承了一个不工作的基因时,就会发生隐性遗传病。如果一个人得到一个工作基因和一个非工作基因的疾病,这个人将是疾病的携带者,但通常不会出现症状。两名携带者的父母同时传递非工作基因,因此,每次怀孕都有一个受影响的孩子,其风险是25%。每次怀孕时,孩子与父母一样是携带者的风险是50%。孩子从父母双方获得工作基因的机会是25%。男性和女性的风险是一样的。
X-连锁遗传病是由X染色体上不工作的基因引起的疾病,主要表现在男性身上。女性的一条X染色体上有一个不工作的基因,就是该疾病的携带者。携带者女性通常不显示症状,因为女性有两条X染色体,只有一条携带非工作基因。男性有一条从母亲那里继承的X染色体,如果男性继承了含有非工作基因的X染色体,他就会患上该病。X连锁疾病的女性携带者每次怀孕都有25%的机会生出像自己一样的携带者女儿,25%的机会生出非携带者女儿,25%的机会生出受疾病影响的儿子,25%的机会生出未受影响的儿子。如果一个患有X连锁疾病的男性能够繁殖,他将把不工作的基因传给他所有的女儿,这些女儿将是携带者。男性不能将X连锁基因传给他的儿子,因为男性总是将其Y染色体而不是X染色体传给男性后代。
X连锁显性疾病是由X染色体上的一个不工作的基因引起的,主要发生在女性身上。患有这些罕见疾病的女性,当她们的X染色体上有某一疾病的非工作基因时,就会受到影响。患有X连锁显性疾病的非工作基因的男性受到的影响比女性更严重,往往不能存活。
CMT疾病的通用命名系统是将其细分为几个主要类型,称为CMT1、CMT2、CMT3、CMT4、CMTX、CMTDI和CMTRI。如果一个负责任的基因已被确定,则在名称上加一个字母,如CMT1A、CMT1B等。最近,人们提出了新的命名方案,根据遗传模式、病因是脱髓鞘性、轴索性还是中间性以及致病基因是否已知来简化这一命名。表1列出了使用通用命名法的CMT类型,并列出了致病基因。一些类型的命名是在遗传原因被充分了解之前,所以这个命名系统存在一些冗余,一些类型被证明不准确地归因于特定的基因,使某些亚型的名称变得过时。由于这些原因,一些名称不再被普遍使用。
CMT1是最常见的CMT形式,其特点是神经髓鞘的异常,导致神经传导速度下降。周围神经髓鞘是由许旺细胞形成的,参与这些许旺细胞形成和功能的基因的异常变化导致了脱髓鞘。CMT1是常染色体显性遗传。CMT1根据特定的基因异常被进一步细分为CMT1A-1F的亚型。CMT1A是迄今为止最常见的单一CMT亚型,占所有CMT病例的50%左右,是由PMP22基因的重复引起。表1中列出了CMT1的其他亚型和负责的基因。
CMT2是该病的一种形式,神经传导速度通常正常或比正常稍慢,但神经信号强度降低。CMT2是由参与轴突结构和功能的异常基因引起的。CMT2主要包括常染色体显性遗传模式。根据特定基因的突变,CMT2又被进一步细分为CMT2A-2Z。CMT2A是最常见的,由MFN2基因的突变引起。表1中列出了CMT2的其他亚型和负责的基因。虽然CMT2是指常染色体显性遗传模式,但一些亚型具有常染色体隐性遗传的特点。
CMT4是严格针对常染色体隐性遗传形式的CMT的分类。CMT4A是由GDAP1基因的异常引起的;然而,当GDAP1基因的损伤性改变足以通过常染色体显性遗传模式传播时,GDAP1基因也是CMT2H和CMT2K的原因。表1中列出了CMT4的其他亚型和负责的基因。SORD相关CMT是目前对由SORD基因变化引起的常染色体隐性轴索型CMT的名称。这个基因最近才被发现是CMT的原因,但可能是常染色体隐性CMT最常见的遗传原因。它尚未在通用的命名系统中得到一个分类。
CMTX是一种由位于X染色体上的基因异常引起的X连锁显性形式的疾病。由GJB1基因变化引起的CMTX1(又名CMT1X),约占CMTX的90%。表1中列出了CMTX的其他亚型和负责的基因。由于GJB1和其他这些引起CMTX的已知和未知基因位于X染色体上,CMTX主要影响男性,然而在CMTX1和CMTX6中,具有X连锁显性遗传的特点,男性比女性受影响更严重。在CMTX1和CMTX6中,受影响的女性通常比男性发病晚,在每个年龄段病情较轻,甚至可能无症状,可能是由于髓鞘雪旺细胞中的X失活。
中间型CMT包括那些具有 "中间 "传导速度的患者,因此不能确定神经病变主要是轴索性还是脱髓鞘性。中间型CMT有显性(CMTDI)和隐性(CMTRI)两种形式。已知的CMTDI和CMTRI亚型的致病基因见表1。
遗传性压迫性神经病变(HNPP)是CMT类疾病中的另一种情况。HNPP是常染色体显性遗传的模式。与CMT1A一样,HNPP是由PMP22基因的变化引起的。与CMT1A不同的是,PMP22基因通常是重复的,而在HNPP中,其中一个PMP22基因被删除,因此HNPP患者不是有两个PMP22的拷贝(来自父母各一个),而是只有一个拷贝。这意味着HNPP患者不能产生足够的PMP22蛋白。
CMT3,也叫德杰林-索塔斯病,已不再是一个常用的名称,因为患有这种病的人已被发现在负责CMT1A、CMT1B、CMT1D或CMT4的基因中出现了基因突变。CMT5和CMT6也是历史上使用的术语,现在归因于MFN2基因的破坏性变化,而MFN2基因现在被认为是导致CMT2A2A的原因。表1中当代亚型名称旁边的括号中包括不再使用的亚型的历史名称。
遗传性感觉神经病(HSN或HSAN)有时也包括在CMT病症组中。某些形式的HSN与CMT的形式有关或相同,而且与造成这些病症的基因有重叠之处。遗传性运动神经病(dHMN)与CMT相似,一些专家认为这些疾病属于同一组。
表1 CMT的亚型、致病基因和遗传模式(改编自Nam,Choi, 2019)
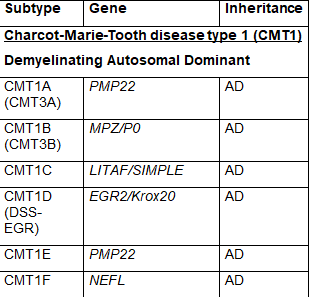
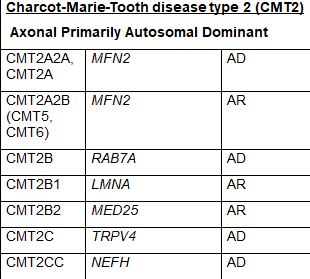

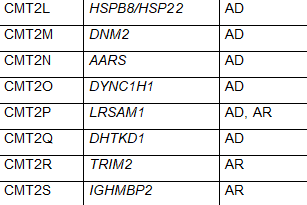
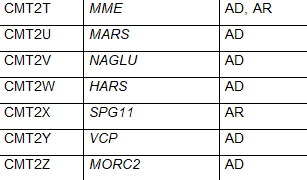
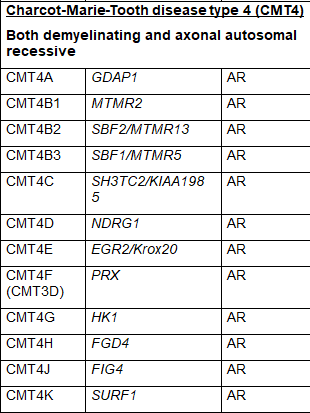


少数 CMT 可有周围神经病以外的其他表现:CMTX1 型可有卒中样发作伴 MRI 白质可逆性病变;CMT5 型伴锥体束征;CMT6 型伴视神经萎缩;CMT7 型伴色素性视网膜炎。
四、流行病学
CMT在全世界所有种族和民族的人中都有发现。流行病学研究对患病率的估计变化很大,部分原因是临床症状和不同疾病形式的差异很大,以及CMT包括哪些疾病的差异。最近的发病率估计在每10万人中有9到28人。CMT的症状通常在青春期、成年早期或中年时期逐渐开始。
CMT 的总体发病率约为 40/100 000,发病率在人种间无明显差别。遗传方式分为常染色体显性遗传、常染色体隐性遗传和 X 连锁隐性遗传等。常见的突变基因包括PMP22、MPZ、GJB1、MFN2。常见的亚型为 CMT1、CMT2、CMTX。CMT1 为常染色体显性遗传的脱髓鞘性 CMT,其中 CMT1A 为最常见的 CMT 亚型(占 40%~50%),其突变基因为 PMP22;CMT1B 占 CMT1 的 3%~5%,其突变基因为 MPZ。X 连锁隐性遗传的 CMTX1 为第 2 常见的 CMT 亚型(占 10%),其突变基因为 GJB1。CMT2 为轴索性 CMT,其中常见的突变基因包括MFN2(占 CMT2 的 20 %)、MPZ(占 CMT2 的 5 %)、NEFL 和 GDAP1 等。CMT4 为常染色体隐性遗传的脱髓鞘性 CMT,其最常见的突变基因为 GDAP1。PMP22 重复、GJB1 突变、PMP22 缺失、MPZ突变、MFN2 突变这 5 种亚型占所有 CMT 的 92%。
五、鉴别诊断
周围神经病是至少100种遗传性综合征的一部分,尽管它通常被其他表现所掩盖。对于哪些疾病与CMT组疾病相似但又是独立的、与CMT组疾病相同或有一些重叠,专家意见不一。
遗传性感觉(和自主)神经病(HSN或HSAN),包括感觉(和不同的自主)神经元/轴受到影响,而运动神经元/轴相对或完全不受影响的情况。神经元表达基因的显性和隐性突变导致HSAN和相关疾病。有超过15种形式的HSAN,它们可以以显性和隐性模式遗传。一些专家认为这些疾病与CMT属于同一组疾病。
远端遗传性运动神经病(DHMN)是运动神经元/轴突受影响但感觉纤维不受影响的情况。这些疾病在临床上和遗传上具有异质性,与远端脊髓性肌肉萎缩症(DMSA)有共同的遗传原因。它们可以以显性或隐性方式遗传。一些专家认为这些疾病与CMT属于同一组疾病。
其他以遗传性神经病变为特征的病症一般被认为不属于CMT组,包括
- 肾上腺髓质神经病(Adrenomyeloneuropathy)
- 多发性神经病、听力损失、共济失调、色素性视网膜炎和白内障(PHARC)。
- 弗里德里希共济失调(Friedreich ataxia)
- NARP - 神经源性肌无力、共济失调和色素性视网膜炎
- 遗传性神经性肌萎缩症(Hereditary neuralgic amyotrophy)
- Refsum综合征
- SCN9A相关的遗传性红斑狼疮症
- Troyer综合征
- 转甲状腺素相关的淀粉样变性疾病(Transthyretin-associated amyloidosis)
- 线粒体神经胃肠脑病(MNGIE)。
有许多远端肌病与CMT有一些共同的特征,但也被认为是在CMT这组疾病之外。这些疾病包括
- 三好氏营养不良症3型(Miyoshi dystrophy type 3)
- 三好早年发病的肌病(Miyoshi early-adult-onset myopathy)
- 远端肌纤维性肌病(Distal myofibrillar myopathy)
- 中段性肌纤维肌病(Mesminopathy myofibrillar myopathy)
- 肌纤维性肌病(Myofibrillar myopathy)
- 野中早年发病的远端肌病(Nonaka early-adult-onset distal myopathy)
- Zaspopathy(Markesbery-Griggs晚期发病的远端肌病)。
- 肌萎缩性侧索硬化症21(以前的MPD2)
- 莱恩早发远端肌病(Laing early-onset distal myopathy)
- 远端肌腱病(Distal myotilinopathy)
- 远端尼布林肌病(Distal nebulin myopathy)
- 韦兰德远端肌病(Welander distal myopathy)
- 远端发病的远端肌病(Distal onset in telethoninopathy)
- 远端肌病(Udd distal myopathy)
- 与CAV3、FLNC和其他基因有关的远端肌病
六、诊断
CMT疾病的诊断可能具有挑战性。诊断的依据是身体症状、家族史和临床测试。临床检查包括神经传导速度(NCV)和肌电图(EMG),前者测量脉冲沿神经传播的速度,后者记录肌肉细胞的电活动。随着分子基因测试的最新进展,对临床诊断为CMT的病人采用缺失复制分析和下一代测序(NGS),大约60%的病人可以找到遗传原因。对于CMT1,56-92%的病人可以找到遗传原因。对于轴索型CMT,17-44%的患者可以找到遗传原因。测试多个基因的样本可以包括7到150个基因,而4个基因的异常(PMP22重复、GJB1、MFN2和MPZ)约占这些基因诊断的90%。

七、治疗
CMT疾病的治疗是对症和支持性的。目前还没有治愈的办法,所以重要的是尽量减少或延缓症状。综合治疗包括物理治疗、鞋子矫正器、腿部支架和手术矫正畸形。补充疗法可能在心理上有所帮助,缓解疼痛和不适,并改善整体生活质量。职业咨询,预测疾病的发展,对年轻患者可能是有用的。遗传咨询在许多方面都有帮助,包括遗传诊断、与研究和支持团体的联系以及对复发风险的了解。
1. 康复治疗 规范的康复治疗能够延缓疾病造成的功能障碍如关节畸形等,维持更好的生活功能和姿态。支具鞋等可改善行走步态。
2. 外科矫形治疗 对于严重的骨骼畸形,特别如高足弓、锤状趾畸形,手术矫形可能有益。
3. 尽量避免使用可能加重 CMT 的药物 如长春新碱、胺碘酮、硼替佐米、铂类、氨苯砜、来氟米特、呋喃妥因、甲硝唑、司他夫定、他克莫司、沙利度胺、扎西他滨等。
4. 遗传咨询与产前诊断 CMT 类型众多,基因确诊后建议遗传咨询,明确病因及家系成员风险。对于严重致残的类型,在家属充分知情、征求意见后,可考虑再次生育时进行产前诊断。
八、罕见病信息登记
如果您愿意寻求不断更新的信息,建议您在此登记患者的信息,即使没有完全确诊,也可以登记,点击进入:
参考资料:
Nam SH, Choi BO/ Clinical and genetic aspects of Charcot-Marie-Tooth disease subtypes. Precision and Future Medicine 2019; 3(2): 43-68.
Vaeth S, Vaeth M, Andersen H, Christensen R, Jensen UB. Charcot-Marie-Tooth disease in Denmark: a nationwide register-based study of mortality, prevalence and incidence. BMJ Open 2017; 7(11): e018048.
Barreto LCLS, Oliveira FS, Nunes PS de França Costa IMP, Garcez CA, Goes GM, et al. Epidemiologic study of Charcot-Marie-Tooth disease: a systematic review. Neuroepidemiology 2016; 46(3):157-165.
Wang Y,Yin F. A review of X-linked Charcot-Marie-Tooth disease. Journal of Child Neurology 2016; 31(6): 761-772.
Foley C, Schofield I, Eglon G, Bailey G, Chinnery PF, Horvath R.Charcot–Marie–Tooth disease in Northern England. Journal of Neurology, Neurosurgery & Psychiatry 2012. 83(5): 572-573.
Bird TD. Charcot-Marie-Tooth (CMT) Hereditary Neuropathy Overview. 1998 Sep 28 [Updated 2021 May 20]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2021. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1358/ Accessed June 29, 2021.
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言






#腓骨肌#
64
#萎缩#
103
#诊断与治疗#
71
#流行病#
85
#肌萎缩#
71