ALK耐药
间变性淋巴瘤激酶(ALK)是肺癌中的一种致癌驱动因子。ALK酪氨酸激酶抑制剂对ALK融合阳性(ALK+)肺癌患者产生显著益处。到目前为止,已有5个ALK TKI获得了FDA的批准,用于治疗晚期ALK+NSCLC(非小细胞肺癌),还有更多正在临床开发中。 尽管ALK+肺癌患者对ALK TKI的反应显著,但几乎所有ALK+肺癌患者最终都会通过在靶和靶外耐药机制复发。临床上在解决耐药机制的异质性和防止疾病复发方面仍然存在挑战。
公众号"医药速览" 发布的这篇文章概述了致癌ALK融合的基本生物学,讨论了目前对ALK导向治疗获得性耐药的理解,并重点介绍了旨在诱导晚期ALK+肺癌长期缓解的最新治疗策略。
小编摘录如下,供专业人士参考了解。
↑ 论文链接:https://doi.org/10.1038/s43018-023-00515-0
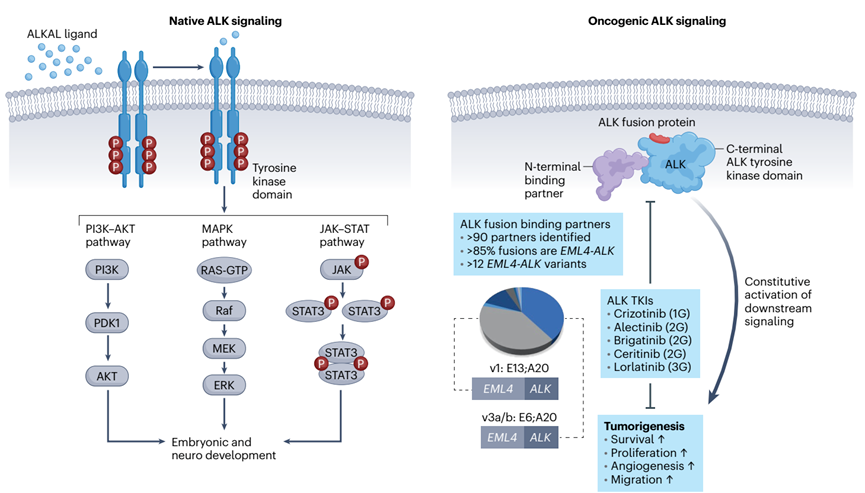
在恶性肿瘤中,ALK点突变或染色体重排导致ALK及其下游信号级联异常激活。在非小细胞肺癌和包括ALCL、弥漫性大B细胞淋巴瘤、炎性肌纤维母细胞瘤、胶质瘤和结肠癌等癌症类型中,致癌的驱动因素是结构性的ALK重排,即ALK的3’端的激酶域编码区与5’端的各种配对基因融合。与天然ALK不同,大多数ALK融合体缺乏跨膜区域并且不锚定在质膜上。ALK已被用作研究肺癌中其他融合癌蛋白的框架,如ROS1、RET、TrkA-TrkC和NRG1。虽然ALK驱动的NSCLC是癌基因成瘾的典型例子,但ALK+NSCLC中的基因组异质性,就结合伙伴、相互易位和断点变异而言,是否解释了对TKIs治疗反应的差异仍不清楚。此外,ALK融合在癌症中的病因很大程度上是未知的,其影响年轻患者的倾向也是如此。
ALK+NSCLC领域已经被不断发展的选择性、有效性和脑渗透性越来越强的ALK TKI所改变,说明了洞察癌症分子依赖性的临床好处。ALK+NSCLC的临床前模型已被证明对于概述ALK的体内外病理生理学和促进ALK抑制剂的开发至关重要。
ALK+肺癌的临床前建模刺激了大量ALK TKI的发展(表1)。Crizotinib是第一代ALK抑制剂,是一种多靶点TKI,最初是作为间充质-上皮转化(MET)的抑制剂而开发的。然而,由于药物对血脑屏障的渗透不良,会观察到中枢神经系统(CNS)的复发。已开发的几种第二代ALK TKIs(其中Ceritinib、Alectinib和Brigatinib已获得FDA批准)在CNS中也显示出临床活性,并克服了一些常见的Crizotinib耐药ALK突变。在没有耐药的ALK突变的情况下,第二代ALK TKI也是有效的,这表明Crizotinib对ALK的不完全抑制。Alectinib、brigatinib和ensartinib在一线试验中直接与Crizotinib进行比较,显示出第二代TKIs在所有方面的卓越疗效。
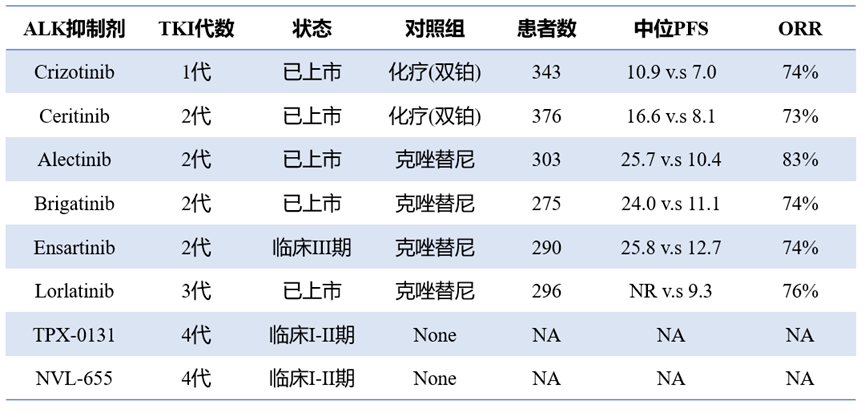
Lorlatinib是第三代大环ALK TKI,设计为高效、选择性和中枢神经系统穿透。临床前研究表明,Lorlatinib对野生型ALK和对第一代和第二代TKI无效的最已知的ALK突变具有很强的活性,在2021年被FDA批准为一线药物。
尽管迭代几代的ALK TKI已取得成功,但仍然受到耐药性的限制。在接受一线Alectinib治疗的ALK+NSCLC患者中,大约一半的患者将在大约2年内经历疾病进展,这是肿瘤演变和出现耐药性的后果。
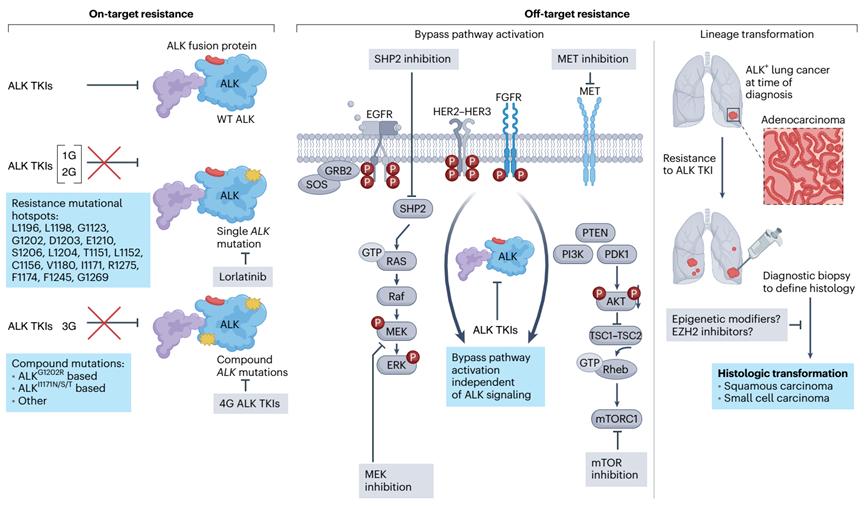
对ALK靶向治疗的耐药性大致可分为ALK依赖和ALK非依赖性。ALK依赖的或“靶点上”的耐药性在很大程度上是由ALK基因中出现单一或复合突变定义的,使肿瘤细胞持续依赖ALK活性。在ALK+肿瘤细胞中,ALK非依赖性或非靶向耐药是通过谱系改变或激活ALK非依赖性信号通路来定义的。 在接受第二代ALK抑制剂治疗的患者中,50-60%的患者通过获得继发性ALK突变而产生耐药性。这些突变普遍存在于TKI结构域中,并通过TKI结合的直接空间位阻、蛋白激酶构象的改变和/或ATP结合的改变而产生耐药性。
在第二代ALK TKI进展的ALK+ NSCLC患者中,大约一半在临床复发时未发现 ALK突变,这表明不依赖于ALK的耐药性,并且下一代ALK抑制剂对该子集仅适度获益患者。赋予ALK TKI耐药性的多种脱靶机制可能发生在患者身上,这使得ALK非依赖性耐药性难以克服。
一类重要的 ALK 非依赖性耐药性是旁路信号的激活,它由遗传改变、蛋白质表达的变化和/或自分泌反馈信号的激活或失调引起。在ALK TKI耐药肿瘤中描述了多个旁路通路,包括 RTK MET71、EGFR72、SRC56、IGF-1R73、HER2和 HER3和KIT72的激活以及下游信号因子MAP2K1的改变、DUSP6、STAT3和NF2。
细胞内信号介质也与获得性耐药有关。致癌ALK信号需要激活MAPK通路。ALK非依赖性MAPK通路重新激活可通过多种机制发生,包括KRAS拷贝数增加、丝裂原活化蛋白激酶激酶1 (MAP2K1) 激活突变或 DUSP6(MAPK 的负调节因子)的缺失。
肿瘤转化为不同的组织亚型与失去对致癌驱动因素的依赖有关,从而导致抗药性。虽然几乎所有新诊断的ALK+NSCLC患者都是腺癌,但在ALK+肺癌患者中,所有代ALK TKI治疗后都发现了小细胞肺癌的转化,尽管转化的频率很低。
由于耐药机制的异质性,ALK TKI 耐药肿瘤的治疗靶向变得复杂。不同的途径可能在一名患者的不同转移灶或一个疾病部位的肿瘤细胞簇中进化,从而导致多克隆耐药性。识别个体患者不同耐药机制的能力越来越强,这可能会造成临床困境,如果有的话,保证治疗靶向,特别是在有多种 FDA 批准或研究药物可用的情况下。
接下来,我们讨论ALK+ NSCLC的治疗策略,这些策略侧重于最大限度地抑制ALK以及克服或预防ALK TKI耐药性的新方法。
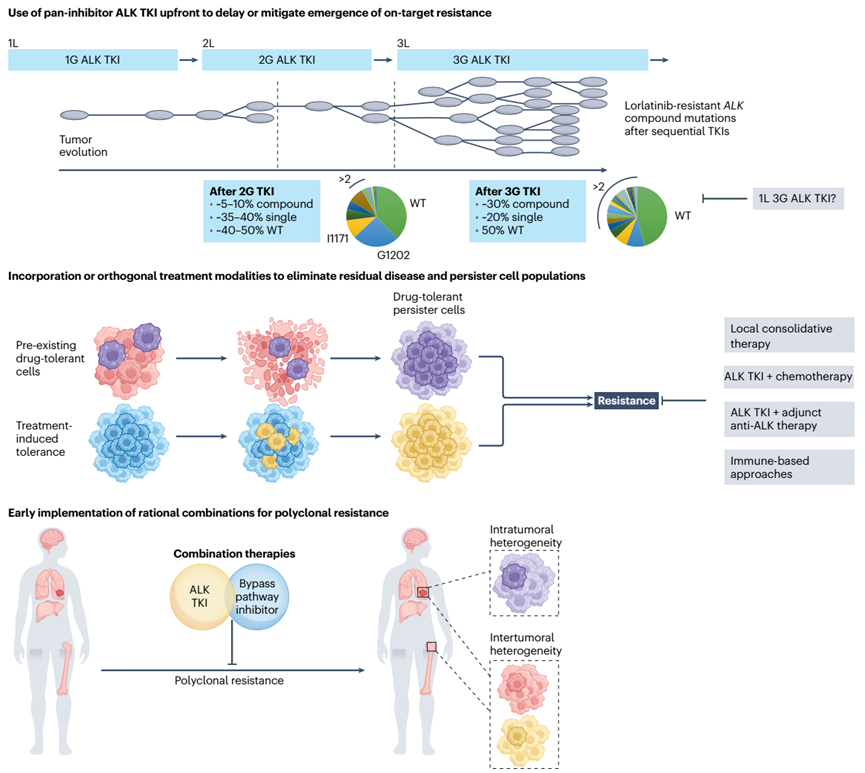
将最有效的ALK抑制剂转移到一线可能会抑制或延迟靶向耐药性的出现并延长反应持续时间。从泛抑制、高效和中枢神经系统渗透性 ALK 开始TKI 以确保最大的细胞减少和反应深度,限制可能出现的肿瘤异质性较弱的第二代 ALK TKI 并延迟靶向耐药性和 CNS 复发。
随着就 ALK TKI 测序达成共识的努力仍在继续,肿瘤携带 ALK 耐药突变的患者可以使用针对该特定突变的 ALK TKI 进行治疗。肿瘤缺乏 ALK 突变的患者可以考虑采用基于 ALK 的组合策略或其他研究方法来解决 ALK 非依赖性耐药性。
由于复合ALK突变,即使在Lorlatinib治疗后,一部分 ALK 驱动的肿瘤仍然对 ALK 成瘾。针对不同类别的复合 ALK 突变可能需要不同的 ALK TKI,而一种新的 ALK TKI 不太可能克服所有靶向Lorlatinib耐药性。
每种 TKI 都可能具有针对 ALK 突变体的独特效力谱,因此可能在具有已知 ALK 依赖性耐药性的下一代 TKI 之后最有用。预计会产生对这些新药产生耐药性的突变,这突出了在 ALK+ NSCLC 中不断追逐耐药性的挑战。从泛单一突变抑制性 ALK TKI开始的一个潜在优势是预防难治性复合 ALK 突变并避免对更高代 TKI 的需求。
序贯 ALK TKI 的使用暴露了日益复杂的靶向耐药突变的问题。因此,在小分子 TKI 之外靶向 ALK 的替代方法值得关注。通过变构或共价抑制或蛋白质降解靶向 ALK 可作为一种补充治疗方法,以规避使用顺序 ALK TKI 暴露的日益复杂的靶向耐药突变。另一种以 ALK 为中心的方法涉及破坏蛋白质-蛋白质相互作用。这些药物是否会在临床上成功开发,以及它们是否会与 ALK TKIs 一起或代替 ALK TKIs 被整合到实践中仍然未知。然而,尽管最大限度地抑制了 ALK,但仍有相当一部分患者会产生不依赖于 ALK 的耐药性,需要替代治疗策略,例如靶向旁路通路或肿瘤微环境因素。
介导ALK TKI耐药的旁路通路的功能特征导致了针对 ALK TKI 复发患者的合理组合方法的开发。迄今为止,组合策略的挑战是毒性的增加会限制每种药物的剂量。另一个潜在的陷阱是治疗的时机,因为组合可能会在阻止耐药性出现方面发挥更大的影响,而不是一旦建立就克服耐药性。
尽管在开始使用 ALK TKI 时通常会出现明显的反应,但即使在治疗稳定几年后,残留疾病通常仍然存在并可能导致复发。越来越多的证据表明,耐药的持留细胞是导致残留疾病的原因。持久状态被认为是可逆的,大多数细胞在药物存在的情况下仍处于细胞周期停滞状态,但一小部分细胞具有重新进入细胞周期的能力。持久性药物可能通过适应性机制破坏 TKI 抑制,包括表观遗传修饰、旁路激活、代谢重编程和改变与肿瘤微环境的相互作用。
在 ALK 驱动的疾病背景下,基于免疫的替代策略可能是更有希望的途径。例如,ALK 代表了疫苗的一个有吸引力的目标,因为它被认为是一种肿瘤抗原,并且在 NSCLC 和 ALCL 中检测到针对它的自身抗体,这表明一些患者可能能够产生自发的抗 ALK 免疫反应。
控制 ALK+ 肺癌谱系可塑性的分子介质及其如何导致 TKI 耐药仍有待确定。需要更多的探索性研究来阐明阻遏物RB1和肿瘤蛋白p53 (TP53)在ALK+ SCLC 转化中的作用以及在发生谱系变化的肿瘤中的相关依赖性。表观遗传景观的调节也正在研究中,作为逆转谱系可塑性和使细胞对 ALK 抑制剂重新敏感的途径。
ALK+肺癌的治疗代表了精准肿瘤学的典范,为适用于各种癌症类型的靶向治疗提供了经验教训。自从在 NSCLC 中发现 EML4-ALK 融合以来,在很短的时间内,ALK 定向疗法的开发取得了巨大进步,从而显着提高了患者的生存率。该领域举例说明了实验室和临床研究的整合如何揭示重要的分子见解,这些见解可以转化为患者的实时临床益处。
然而,转移性ALK+ 肺癌仍然无法治愈,该疾病的治疗仍面临许多挑战。尽管复发时临床样本的基因分型为耐药生物学提供了信息并导致了新一代ALK TKI 的发展,但它也揭示了越来越多的TKI难治性 ALK 耐药突变和异质性脱靶 ALK 逃逸机制。阐明耐药性的分子驱动因素并继续开发创新疗法以克服耐药性至关重要。
更深入地了解ALK+肺癌的独特生物学特性和治疗的持续进展,最终将催生一波又一波的转化研究工作,致力于实现延长ALK+肺癌患者生命和诱导治愈的总体目标。
参考文献:
Schneider, J.L., Lin, J.J. & Shaw, A.T. ALK-positive lung cancer: a moving target.Nat Cancer 4, 330–343 (2023). https://doi.org/10.1038/s43018-023-00515-0
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言