疑难病例分享|腹痛伴脾大:一例不明原因肝功异常
2019-03-28 小楠 中国医学论坛报
近年来,中国肝脏病学进入了快速发展阶段,肝病学的学术水平有了很大提高,但与发达国家相比,仍存在一定差距。临床思维作为疾病诊治中至关重要的一环,能体现出临床医生在诊治中的理论与实践融会贯通的程度。因此,对于我国肝病领域的临床医生来说,培养和提升清晰的临床诊疗思维非常重要。
近年来,中国肝脏病学进入了快速发展阶段,肝病学的学术水平有了很大提高,但与发达国家相比,仍存在一定差距。临床思维作为疾病诊治中至关重要的一环,能体现出临床医生在诊治中的理论与实践融会贯通的程度。因此,对于我国肝病领域的临床医生来说,培养和提升清晰的临床诊疗思维非常重要。基于此,在3月17日第三届华润三九全国肝病高峰论坛上,四川大学华西医院吴东波博士分享了一例肝脏疑难病例的病因诊断过程。并随后在现场与点评专家在一问一答中进行思维、观点的碰撞,并促进现场与会医生互相启发,相互学习。现将病例内容整理如下,与广大读者分享。
病例作者

四川大学华西医院 吴东波博士
病史简介
患者:男性,16岁;主诉:因“反复乏力、皮肤、巩膜黄染一年以上。”于2017-11-15入院。身高:165 cm;体重:55 kg;BMI:20.2。
现病史:患者于1年前受凉后出现畏寒、发热及鼻塞、流涕,经治疗后好转,但逐渐出现皮肤黄染及上腹胀痛,伴恶心、呕吐,呕吐为胃内容物。后患者就诊于重庆某医院行相关检查诊断为“急性黄疸型肝炎”。予保肝退黄等治疗,稍好转后出院。院外给予多烯磷脂酰胆碱、熊去氧胆酸、复方甘草酸苷等药物治疗,效果不佳,黄疸持续存在,TBIL水平在50~100 μmol/L。
2个月前,当地医院考虑患者的临床诊断为“自身免疫性肝炎”,予加用强的松30 mg/qd,硫唑嘌呤50 mg/qd,患者家属自述患者的病痛无明显改变,出现颜面毛发增粗,色素沉着。
1周前患者再次出现恶心、 呕吐、剧烈腹痛、呕吐咖啡色胃内容物两次,约150 ml/次,在当地医院行腹部平片示:结肠积气扩张、内见大量气体粪便影,考虑诊断“不完全性肠梗阻,应急性溃疡出血”,予停用强的松,并禁食、保肝、护胃、抗感染等对症处理,患者自动出院。(补充病史:尿卟啉阴性)
2天前就诊于我院急诊。全腹CT示:盆腔少量积液,横结肠、升结肠部分肠腔稍积气扩张,回盲部结构稍模糊,阑尾显示欠清,腔内少许粪石影可能,临近肠系膜上淋巴结多发显示。肝脏实质密度稍降低,轻度脂肪肝可能,胆汁分层,脾脏增大。
既往史、个人史:无肝炎、结核或其他传染病史,无药物过敏史,无吸烟及嗜酒史,无外伤史。患者自述从小皮肤对光过敏。
家族史:家族中无类似患者,父母及兄长健康。
入院查体:T 36.30℃,P 128次/分,R 22次/分, Bp 127/96 mmHg。神志清楚,慢性病容,皮肤巩膜轻度黄染,颜面毛发增多,色素沉着,痤疮散在分布。右侧季肋区黑毛痣样胎记,形状不规则,直径约15 cm。心律齐,各瓣膜区未闻及杂音。双肺呼吸音清,未闻及干湿罗音。腹部外形正常,全腹软,无压痛及反跳痛,肝脾肋下未触及,双下肢无水肿。
入院前辅助检查:TBIL 95.1μmol/L,DBIL 77.1μmol/L,ALT 146 IU/L,AST 104 IU/L,ALP 118 IU/L,GGT 280 IU/L。
入院情况:青年男性,起病缓,病程长。肝功异常以胆红素升高为主。伴有腹痛症状和皮损表现,CT提示肝脾长大。
入院诊断
①黄疸待诊:卟啉病?进行性家族性肝内胆汁郁积症?自身免疫性肝病?其他;②不完全性肠梗阻;③应激性溃疡;④脾脏长大。
入院后检查
大便常规正常;尿常规:PRO 0.1(+/-),尿胆红素(2+);血常规:HGB 100 g/L,PLT 237×109/L,WBC 7.28×109/L;生化:TBIL 113.9 μmol/L,DBIL 94.3 μmol/L,ALT 180 IU/L,AST 128 IU/L,TBA 95.5 μmol/L,ALP 141 IU/L,GGT 382 IU/L;血氨:42 μmol/L;AFP/CEA/CA125:正常;肝炎全套阴性;TP/HIV阴性;TORCH检查正常;EB DNA及CMV DNA阴性;直接抗人球蛋白试验阴性;ANA及自免肝相关抗体阴性,免疫全套正常;IgG、IgM、IgG4和铜蓝蛋白正常;甲状腺功能正常;FE、总铁结合力、血清铁饱和度正常;凝血功能:PT 13.9s,INR 1.18。
上腹部MRCP:动脉早期肝右前叶高信号影,胆囊结石,脾脏增大,扫及右侧胸腔积液。
肝穿刺活检:
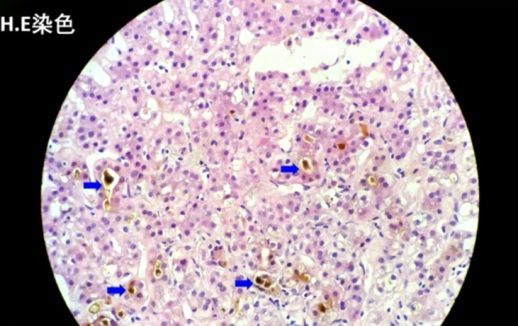
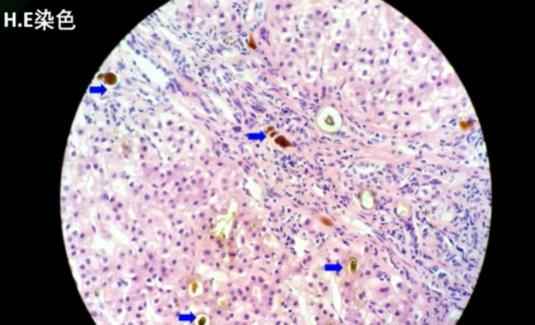
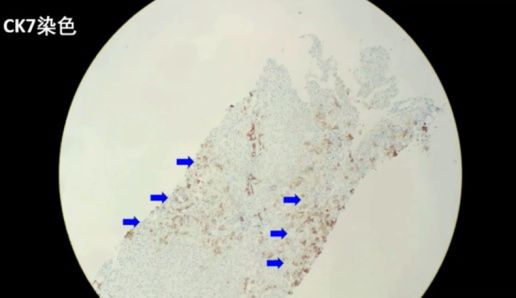
病理结果:共查见约9个门管区。肝细胞胆汁淤积,可见毛细胆栓形成(以腺泡3区为重,该区d-PAS阳性的Kupffer细胞增生)。肝小叶内见少数点灶状坏死,界面欠完整。门管区一些淋巴细胞、单核细胞及中性粒细胞浸润,CK7示小胆管增生,少部分肝细胞呈胆汁淤积模式。Foot及Masson染色示纤维组织增生,门管区扩大,3处可见纤维隔形成。
免疫组化:HBsAG(-)、HBcAG(-)、IgG4(-)。特殊染色:PAS、铜染色、铁染色未见明显异常。
病理诊断
胆汁淤积性肝病(以腺泡3区为重),纤维化评分相当于2~3。
进一步分析患者症状的原因有哪些?
①肝功异常原因:嗜肝病毒、非嗜肝病毒;铜代谢;甲状腺功能;抗核抗体、自身免疫性肝病系列,药物损伤,化学物质损伤。结合病史、免疫学指标、影像学、病理学检查。
②腹痛的原因:腹部CT发现肠梗阻、无肠肿瘤、查体无腹膜炎改变。
③腹痛伴肝功异常:中毒:铅中毒;自身免疫性疾病:过敏性紫癜;内分泌疾病:甲状腺亢进等;寄生虫感染:寄生虫移行;遗传代谢性肝病:血色病,wilson病,PFIC,卟啉病等。
结合病史及患者症状,患者卟啉病可能性大。
基因检测

FECH基因突变:
①FECH基因报道与红细胞生成原卟啉症相关。疾病报道遗传方式为常染色体隐性遗传(AR);
②UROD基因报道与卟啉迟发性皮肤易感性相关。疾病报道遗传方式为常染色体显性或隐性遗传(AD/AR)。
最终诊断
①血卟啉病(红细胞生成性原卟啉病);②不完全性肠梗阻;③应激性溃疡;④脾大。
临床随访:近期电话联系,患者病情稳定。
复查结果示:TBIL 18.6 μmol/L,DBIL 11.4 μmol/L,ALT 356 IU/L,AST 206 IU/L,TBA 13.2 μmol/L,ALP 165 IU/L, GGT 329 IU/L。
诊疗体会
①对于不明原因肝功异常患者需要结合患者临床特点及实验室检查结果进行综合分析。②不明原因肝功异常患者中,肝活检病理组织学检查有助于明确诊断,进一步缩小鉴别诊断范围。③对于诊断遗传代谢性肝病,分子及基因检测非常重要。
专家讨论

左上至右下:赵景民教授、潘晨教授、谢雯教授、曾义岚教授、何方平教授
解放军第五医学中心赵景民教授、福州市传染病医院潘晨教授、首都医科大学附属北京地坛医院谢雯教授、成都市公共卫生临床医学中心曾义岚教授和新疆医科大学第一附属医院何方平教授就病例内容分别提出了自己的见解和分析,现场讨论气氛热烈。同行间的思维碰撞、专家思路点拨对于提升医生临床判断能力有积极的作用。现综合点评专家对该病例的主要观点,分享如下:
血卟啉病由Stokvis于1889年首次报道,是一种可累及神经系统或(和)皮肤的代谢性疾病。属少见病,为常染色体显性或隐性遗传疾病。血红素合成途径中PBG脱氨酶(尿卟啉原合成酶)缺乏所致卟啉代谢紊乱而发生的疾病。临床表现主要有光感性皮肤损害、腹痛及神经精神症状。
卟啉病分类:
临床上常见类型有:急性间歇性血卟啉病(AIP)、迟发性皮肤性卟啉病(PCT)、红细胞生成性卟啉病(EPP)。
EPP是一种常染色体显性遗传病(可有AR),突变基因是亚铁螯合酶(FECH)基因,导致线粒体铁螯合酶的缺陷,引起原卟啉在红细胞、血浆、肝脏和皮肤的蓄积,出现相应的临床症状。主要表现是幼儿时期即出现的痛性皮肤光敏性红斑。肝胆系统受累不多见,发生在1%~4%EPP患者中,表现为胆汁淤积和慢性肝病,严重者可导致肝功能衰竭。FECH是卟啉代谢过程中的第8个酶,催化铁与光卟啉Ⅸ结合形成血红素,EPP患者FECH的活性仅为正常人的10%~25%,FECH的缺失使原卟啉大量沉积在红细胞内,随着红细胞的老化释放到血浆中。原卟啉可由肝排入胆汁和粪便中,因此EPP患者红细胞、血浆和大便中原卟啉增加,而尿卟啉阴性。
肝组织病理特点:HE染色光镜下可见肝细胞内、肝细胞间微单管内、肝窦内和巨噬细胞胞浆内棕黄色颗粒样沉积物沉积,这些沉积物实为原卟啉在肝脏内的沉积。偏光显微镜下具有双折光特性,呈现为“Maltese”十字样。
治疗:尚无明确的特效药物治疗。避免光照,如衣物、防晒霜等。避免饮酒和吸烟。使用新药物前查询有无诱发可能。急性期临床对症处理。包括:控制感染、控制心率、纠正水电解质平衡紊乱如低钠血症等,保证能量供应尤其是碳水化合物的补充,葡萄糖能抑制ALA合成酶活性,急性发作时5%~10%葡萄糖静脉滴注,配合高糖饮食能迅速缓解症状(腹痛缓解明显)。补充胡萝卜素,推荐剂量每日口服60~180 mg,维持血清浓度4~6 mg/L(约1/3的人有效)。可试用考来烯酸和熊去氧胆酸(阻断原卟啉的肠肝循环、促进原卟啉从粪便中排出)反复输血提供血红素、血浆置换(清除过多的原卟啉来治疗EPP合并急性胆汁淤积)。
晚期进行肝移植。及其他新的治疗方式。
卟啉病诊断要点:
发作性腹痛,腹部检查无明显压痛;神经精神症状;皮肤损害;血/尿卟胆原阳性;小便在太阳暴晒下颜色变红。和胆汁淤积不同,原卟啉在肝细胞内的沉积,在偏振光显微镜下折光性很强。另外,针对此病例的诊断还有必要补充一些工作:①需补充电镜下结构(冰晶样结构)。②进一步完善基因检测,因为其他疾病也可能有FECH基因的突变。卟啉病是罕见病,在诊断时不应作为一线考虑,应该放在后线。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言






#疑难病例#
114
很好的学习机会
180
很好的学习机会
1
很好的学习机会
116
很好的学习机会
177
挺详细的病例
104
#腹痛#
73
学习了很有用不错
62
谢谢了,学习
66
谢谢分享
70