
专家论坛|甘梅富:原发性胆汁性胆管炎的病理学诊断
2024-07-05 临床肝胆病杂志 临床肝胆病杂志 发表于上海

本文就原发性胆汁性胆管炎的临床病理特征及治疗进展做一概述。
原发性胆汁性胆管炎(PBC),原称为原发性胆汁性肝硬化,是一种非化脓性慢性自身免疫性肝内胆汁淤积性疾病。其发病机制目前认为与多种遗传易感性和环境触发因素导致免疫紊乱有关。PBC发病机制中的标志性事件是针对丙酮酸脱氢酶复合物E2亚单位(PDC-E2)的免疫耐受缺失,最终导致胆管上皮细胞损伤。如不给予规范治疗,PBC最终会进展为肝纤维化、肝硬化及肝癌,甚至死亡。
PBC呈全球性分布,可发生于所有种族。目前大多数关于PBC的流行病学数据来自欧洲,报告了发病率和患病率分别为每10万居民0.3~5.8和1.9~40。由于亚太地区监测的改善和报告的增加,PBC的发病率和流行率呈增加趋势。PBC发病年龄以40~50岁多见,25岁以下少见。目前认为男女发病率之比为1∶4~1∶6,而不是之前普遍认为的1∶10。Fan等在中国迄今为止最大规模的研究中发现,2001—2016年PBC患者人数显著增加,女性与男性的发病比率为6.1∶1,与韩国研究的6.2∶1及日本7∶1非常相似。相比女性PBC患者,男性PBC患者在确诊时年龄更大,血清碱性磷酸酶(ALP)及胆红素水平更高,往往处于更高的疾病进展期,对熊去氧胆酸(UDCA)应答不佳,继发原发性肝细胞癌风险亦更高,整体预后更差。
1发病机制
几项针对不同患者群体的病例对照研究已经确定了PBC与特定环境的关联。其中最一致的是,PBC与吸烟和尿路感染有关,这种关联可能部分解释了该疾病以女性为主的原因。自身抗原PDC-E2易受环境改变的影响,特别是在某些感染或暴露于化妆品、香水和食用色素中的某些化学物质之后。人类PDC-E2与大肠杆菌PDC-E2相似,特别是在AMA免疫优势表位区。这种分子模仿与疾病的耐受性破坏和发病机制有关。据推测,在PDC-E2耐受破坏机制下,嗜芳烃新鞘氨醇菌、贝塔病毒和德氏乳杆菌可能是PBC的潜在诱因。PBC具有遗传易感性,且有明显家族聚集现象。已知病例的一级亲属中PBC的患病率估计在1.1%~6.4%,远高于一般人群。只有一例PBC阳性亲属的家庭患病率为4.3%。
据报道,PBC患者的后代患PBC的风险高于一般人群(1.2%),女儿的风险甚至更高(2.3%)。人类白细胞抗原(HLA)位点被认为与PBC易感性以及各种其他自身免疫或免疫相关疾病密切相关。在包括日本和中国在内的亚洲人群中,HLA-DRB1*08∶03为易感等位基因(OR:1.77~3.17),HLA-DQB1*03∶01、HLA-DQB1*06∶04和HLA-DRB1*13∶02为保护性等位基因(OR:0.19~0.54)。有国际PBC研究小组进行了最大的全基因组荟萃分析(Meta-GWAS),结合了来自5个欧洲(10 516例)和2个东亚(20 772例)队列前后报道的基因型数据鉴定出23个新的PBC易感基因位点(常染色体上的FCLR3、CACNA1S、LINC00299、DNMT3A、TMEM163、RARB、TET2、ST8SIA4、NDFIP1、ATG5、CCR6、ITGB8、ZC3HAV1、MYC、ANP32B、WDFY4、DEAF1、ETS1、SRP54、RIN3、DPEP2和CD226,X染色体上的GRIPAP1)。在这些PBC易感位点中,大约60%仅在欧洲人群中被发现,而CD28、CCR6和TNFSF15仅在东亚人群中发现。截至2022年12月,全基因组关联研究(GWAS)和相关的荟萃分析在包括欧洲与东亚在内的各种人群中确定了大约70个PBC易感基因位点。这些基因主要涉及4种主要疾病途径:(1)HLA的抗原呈递;(2)白细胞介素-12相关途径;(3)细胞对肿瘤坏死因子的反应;(4)B淋巴细胞活化、成熟和分化途径。最近报道了利用GWAS鉴定的疾病易感基因对PBC等几种疾病进行计算机药物疗效筛选的研究,该方法可以利用药物数据库、治疗靶点数据库和蛋白-蛋白相互作用数据库,识别出与疾病易感基因蛋白产物存在蛋白-蛋白相互作用的候选药物分子靶点。除了用于每种疾病的药物之外,这种方法还可以潜在地识别以前未开发的药物分子靶点。
2临床特点
PBC大多隐匿起病,进展较缓慢。约1/3患者可长期无任何临床症状。当出现症状时,疲乏感是报告最多的主诉(50%~78%的患者)。抗线粒体抗体(AMA)阳性是该病的标志,也被认为在PBC发病机制中起重要作用,因为针对线粒体的自身抗体会导致乏力。在较早的研究中,PBC患者出现皮肤瘙痒为20%~70%。PBC患者可出现脂溶性维生素吸收不良。维生素A缺乏可表现为眼部症状(夜盲症)和毛囊角化病。
维生素D缺乏可能导致继发于缺钙的骨质疏松症和骨软化症,在极端情况下可能表现为感觉异常和手足搐缩。维生素K缺乏可能导致血肿和出血的风险增加。随着疾病进展,可出现其他胆汁淤积以及肝硬化相关的并发症和临床表现。PBC常与其他自身免疫性疾病伴发,可有相应临床症状。
PBC血清生化检查以ALP和GGT升高为主要特征,血清ALT和AST正常或轻度增高。随着疾病进展血清胆红素逐渐增高,以结合胆红素升高为主。免疫学检查表现为高γ球蛋白血症,免疫球蛋白(Ig)M升高,IgG及IgA正常或轻度升高。据报道90%~95%的PBC患者AMA阳性,是本病典型的免疫学指标,但AMA是否阳性与PBC的预后或临床转归无关。5%~15%的PBC患者AMA阴性,又称为自身免疫性胆管炎,临床特征及病理学特征类似。然而,AMA阳性也可以见于各种肝内和肝外疾病,如自身免疫性肝炎(AIH)、丙型肝炎、各种病因引起的急性肝衰竭、系统性红斑狼疮、Sjögren综合征(干燥综合征),以及慢性细菌感染。AMA阳性也存在于一般人群中,在意大利的发病率为0.5%,日本的一项包含1 714例健康受试者的研究中为0.64%。在一项研究中,AMA阳性人群PBC 5年发病率为16%。在其他自身抗体方面,50%~56%的PBC患者抗核抗体呈阳性。PBC特异性抗核抗体分为核点型(如抗gp210抗体)和核周型(如抗sp100抗体),这些抗体的总体阳性率在PBC患者血清中不高,但在AMA阴性患者中可达85%,且具有较高的特异性。Yang等发现,我国PBC患者中抗gp210抗体阳性率为48.2%,且与预后相关;抗gp210抗体高滴度的患者胆汁淤积和肝损伤程度更重,并且通常与更严重的界面性肝小叶炎有关。抗gp210抗体还与治疗应答相关,UDCA治疗有效者的gp210抗体水平明显降低。抗sp100抗体诊断PBC的特异性高达94%~98%,对诊断AMA阴性者具有重要意义。2017年欧洲肝病学会发布的PBC指南建议,抗sp100抗体和抗gp210抗体双阳性的AMA阴性患者可诊断为PBC。sp100抗体在我国PBC患者中的阳性率约为14.1%。
3病理学特点
基本病理改变为:汇管区胆管非化脓性炎性损害,汇管区炎症,淋巴细胞及浆细胞浸润,胆管损伤,胆管基底膜破坏,胆管上皮细胞排列不整,胞浆嗜酸性,肉芽肿形成(图1);界面炎通常较轻。早期汇管区周围带细胆管反应性增生(图2);后期胆管结构逐渐消失(图3);随疾病进展,汇管区可出现纤维化,并逐渐出现汇管区-汇管区桥接纤维化,周边肝细胞呈慢性胆盐淤积性改变,最终形成胆汁性肝硬化(图4)。
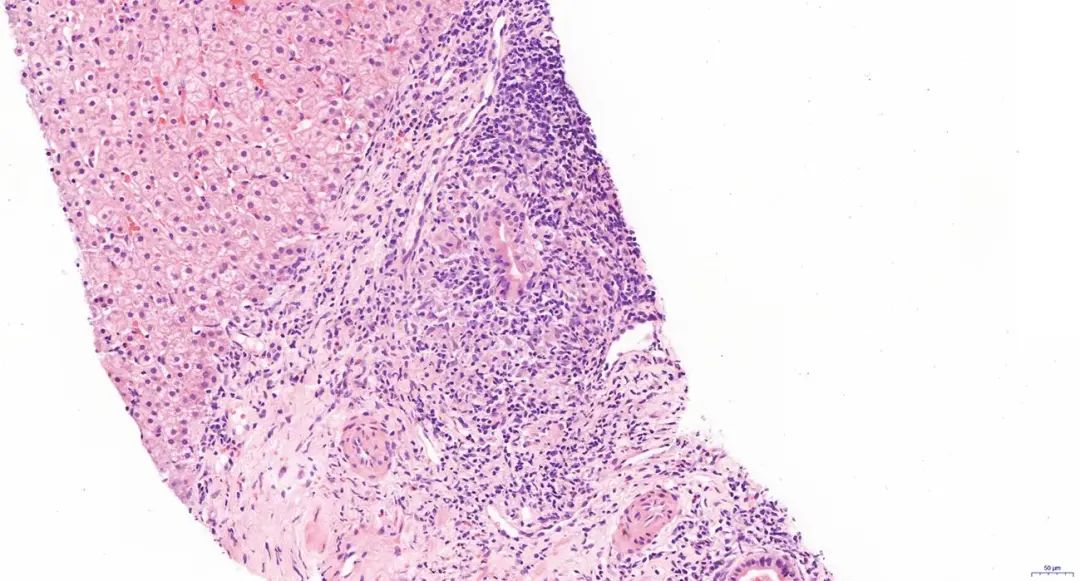
注:汇管区较多淋巴细胞、浆细胞浸润,胆管上皮细胞排列不整,胞浆嗜酸性,肉芽肿形成(HE染色,×20)。
图1 PBC Ⅰ期
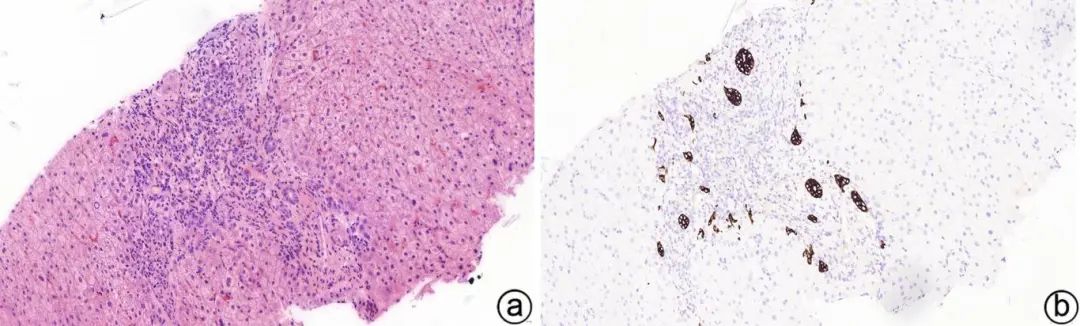
注:a,汇管区周边细胆管增生,可见界板性肝炎及炎性纤维间隔(HE染色,×20);b,CK7免疫组化染色显示反应性增生的细胆管(EnVision法,×20)。
图2 PBC Ⅱ期
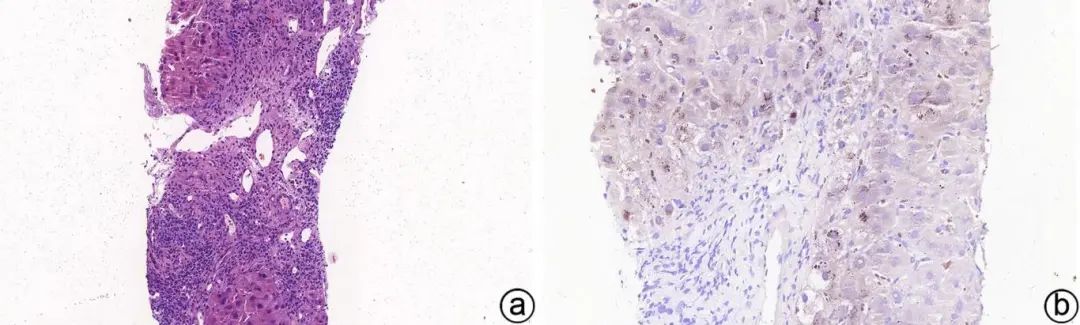
注:a,汇管区固有小胆管缺失,汇管区及间隔周围界面炎范围扩大,可见桥接性纤维化(HE染色,×20);b,汇管区周边肝细胞内铜颗粒沉积(罗丹宁染色,×40)。
图3 PBC Ⅲ期
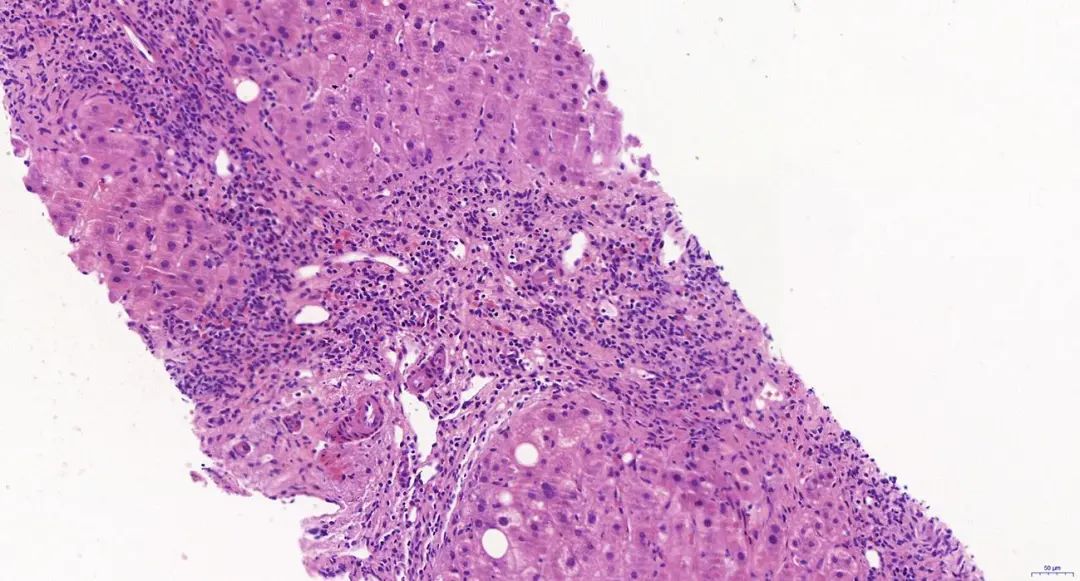
注:汇管区周边肝细胞体积增大,胞质疏松,呈羽毛状变性,形成汇管区-汇管桥接性纤维化(HE染色,×20)。
图4 PBC Ⅳ期
组织学分期(Ludwig分期系统):(1)Ⅰ期,非化脓性胆管炎期:炎症局限于汇管区,具有特征性旺炽性胆管病变。损伤小胆管周围以小淋巴细胞为主,可见淋巴滤泡,可见较多浆细胞,散在嗜酸性粒细胞,偶见少量中性粒细胞,可见胆管反应。约50%的病例损伤小胆管周围可见中央无坏死的肉芽肿。小叶内早期一般不见淤胆,病变较轻。(2)Ⅱ期,细胆管增生期:汇管区固有小胆管数目减少,炎症突破界板,累及小叶内,形成界板性肝炎及炎性纤维间隔,汇管区周围细胆管增生。汇管区及间隔周围肝细胞内可见胆盐淤积。(3)Ⅲ期,纤维化期:汇管区多数小胆管缺失,汇管区及间隔周围界面炎范围扩大,扩大的汇管区相连,可见桥接性纤维化。汇管区及间隔周围肝细胞胆盐淤积明显。(4)Ⅳ期,肝硬化期:除上述改变以外,可见可疑肝硬化或明确肝硬化表现。PBC个汇管区病变可不一致,同一病例可同时见到各期病变。
免疫组化染色CK7和CK19可以提示小叶间固有小胆管损害或数量减少,并可以显示汇管区周围细胆管增生。CK7可显示汇管区周围肝细胞胆管化。特殊染色铜染色可显示汇管区周边肝细胞铜颗粒沉积。电镜可观察到胆管上皮损伤,肉芽肿结构,汇管区周边肝细胞内含铜溶酶体。
鉴别诊断:(1)原发性硬化性胆管炎:多见于男性,血清学AMA阴性,pANCA(80%)阳性,影像学提示胆管呈串珠样改变。镜下表现多累及大胆管,胆管周围硬化性改变(洋葱皮样),炎症反应不显著,通常为淋巴细胞,浆细胞少见,无肉芽肿结构。部分患者可伴发溃疡性结肠炎。(2)肝外胆管阻塞:无血清学异常,影像学表现为肝内胆管扩张。镜下表现为急性胆管炎,胆管增生、扩张,间质疏松水肿,炎细胞主要为中性粒细胞,无肉芽肿。(3)结节病:汇管区无胆管上皮显著炎性损伤,无致密淋巴细胞、浆细胞浸润,肉芽肿形态更好,数量更多,肝小叶中也存在肉芽肿。同时可见其他系统受累的证据。(4)结核:肉芽肿结构与胆管上皮不密切,肉芽肿更大,可见坏死。抗酸染色阳性(阳性率不高),结核分枝杆菌DNA检测阳性。(5)药物所致胆管损伤:小叶内可见活动性炎症性损害,有中性粒细胞,无致密淋巴细胞、浆细胞浸润,肝小叶中央静脉周围带见融合性坏死。临床有用药史。(6)自身免疫性肝炎:无显著胆管上皮损伤表现,重度界面炎,肝细胞内见淋巴细胞穿入现象,小叶内炎症和玫瑰结形成,中央静脉周围融合坏死。(7)慢性病毒性肝炎:组织学及血清学有病原学证据,转氨酶升高为主,ALP、GGT升高不明显,AMA阴性。有时可见胆管损害及致密淋巴细胞浸润(如慢性丙型肝炎),一般无肉芽肿及慢性淤胆表现。
部分自身免疫性肝病患者同时或相继出现AIH和PBC的血清学及组织学特征(图5),称为PBC-AIH重叠综合征。国际上多采用“巴黎标准”诊断PBC-AIH重叠综合征。至少符合每种疾病的3个公认特征中的2个即可做出诊断,其中AIH组织学改变是必需条件。AIH的3个特点:(1)血清ALT水平的5倍以上正常值上限(ULN);(2) IgG水平>2×ULN或抗平滑肌抗体阳性;(3)中重度界面肝炎的组织学特征。PBC的3个特点:(1)血清ALP水平>2×ULN或GGT>5×ULN或更高;(2)AMA阳性;(3)旺炽性胆管病变的组织学证据。《原发性胆汁性胆管炎的诊断和治疗指南(2021)》推荐意见为PBC患者若同时符合以下3项标准中的2项,即可诊断为PBC-AIH重叠综合征:(1)肝组织学提示中重度界面性肝炎(必需);(2)ALT/AST≥5×ULN;(3)IgG≥1.3×ULN或抗平滑肌抗体阳性。
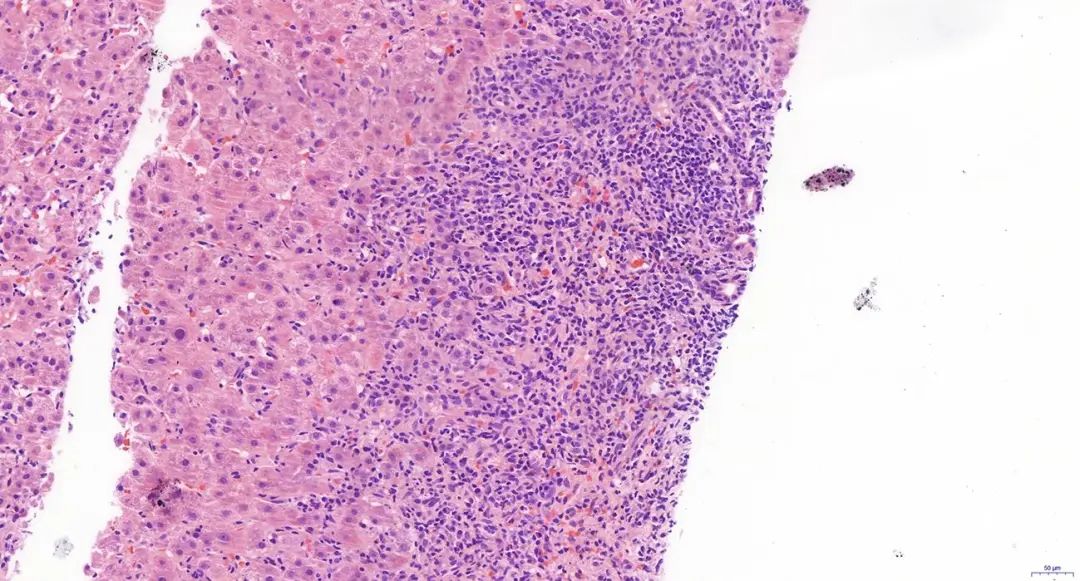
注:汇管区大量淋巴细胞、浆细胞浸润,中重度界面炎,可见旺炽性胆管病变及肉芽肿(HE染色,×20)。
图5 PBC-AIH重叠综合征
4小结与展望
近年来对PBC中胆道损伤病理生理学机制的深入了解,以及针对不同分子靶点新药的研发,使得风险预测模型在PBC中的作用越来越重要。在疾病进展早期决定因素证据的推动下,目前疾病风险分层从评估治疗1年后UDCA应答情况提前至疾病确诊开始,进而及时监测和调整治疗方案进行个体化精准治疗。通过整合新的生物标志物,进一步改进预后模型将成为可能,这些生物标志物提供了多层信息,这些信息与疾病过程的发病机制直接相关。未来的诊断及预后模型内将纳入多层次包括遗传学、表观遗传学、分子和代谢物改变等涉及发病机制的多个方面的组学信息,结合新药的研发,PBC的诊治将进入新阶段。
全文下载
https://www.lcgdbzz.org/cn/article/doi/10.12449/JCH240604

本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言












#病理学# #活组织检查# #原发性胆汁性胆管炎#
67