Duchenne型肌营养不良1例
2020-05-15 杜娟 刘星鹭 陈 诚 国际检验医学杂志

Duchenne型肌营养不良( DMD)亦称假肥大型肌 营养不良症,是一类X连锁隐性致死性遗传疾病,是 肌营养不良蛋白基因突变引起的肌营养不良蛋白功 能缺失或不足,导致肌肉无力,运动迟缓,行走丧失,
Duchenne型肌营养不良( DMD)亦称假肥大型肌 营养不良症,是一类X连锁隐性致死性遗传疾病,是 肌营养不良蛋白基因突变引起的肌营养不良蛋白功 能缺失或不足,导致肌肉无力,运动迟缓,行走丧失, 呼吸障碍和心肌病等[ 1]。现报道1例 DMD诊断 过程。
1 病例资料
患儿,男, 3岁,因“肝功能异常6月余”入院。患 儿于6月前体检发现肝功能异常,血常规白细胞计数 轻度增高,尿液巨细胞病毒DNA阳性,以“巨细胞病 毒肝炎、急性上呼吸道感染”住院,临床予更昔洛韦抗 病毒治疗,护肝治疗,同时予转移因子口服液调节免 疫治疗,直至巨细胞DNA转阴后出院。后间断复查 肝功均异常,谷草转氨酶( AST)及谷丙转氨酶( ALT) 均维持在500 U/L左右。患儿精神、食欲、睡眠尚可, 大小便正常。其父母和16岁哥哥正常,无类似家族 史。既往健康良好,走路易疲劳,父母非近亲婚配,母 亲妊娠、分娩过程无异常,患儿出生至发病时无传染 病、无食物及药物过敏史。入院体格检查:小腿围 22. 1 cm,生命体征平稳,神志清楚,精神可,颈软,咽 无充血,双侧扁桃体无大,全身淋巴结无肿大,双肺呼 吸音粗,未闻及干湿性啰音,心音有力,律齐,无杂音, 腹软,无压痛及反跳痛,肝脾肋下未及,双下肢无水 肿。肠鸣音可,四肢活动正常。生理反射存在,病理 反射未引出。双侧腓肠肌明显肥大,触之有坚实感, 弹性欠佳。仰卧位时起立单侧手撑地后站立( Gowe r s 征阳性)。下阶梯及快步行走时呈典型鸭步。实验室 检查: ALT 610 U/L,AST 460 U/L,肌酸激酶( CK) 35 750 U/L,肌酸激酶同工酶 MB( CK-MB) 12 310U/L,血清α -羟丁酸脱氢酶( α -HBDH) 1 806 U/L,乳 酸脱氢酶( LDH) 2 564 U/L。甲肝、乙肝、丙肝、戊肝 抗体检查正常。肝胆脾胰心脏超声未见明显异常,左 心功能正常。肌电图:右上肢三角肌、右下肢股四头 肌静息状态见异常自发电活动,考虑肌源性损害可能 性大。家属签署知情同意书后,取患儿外周血进行 DMD基因检测:MLPA未检测到DMD基因外显子 大片段缺失/重复;基因测序共检测到6个变异,其中 5个为单核苷酸多态性,而外显子22的无义变异 c. 2941G>T p. Gl u981Te r 未见文献报道,但会导致 蛋白编码提前终止。诊断: Duchenne型肌营养不良, 因该病无特效药物治疗,患儿一般情况尚可,与家属 沟通后办理出院。见图1、 2。
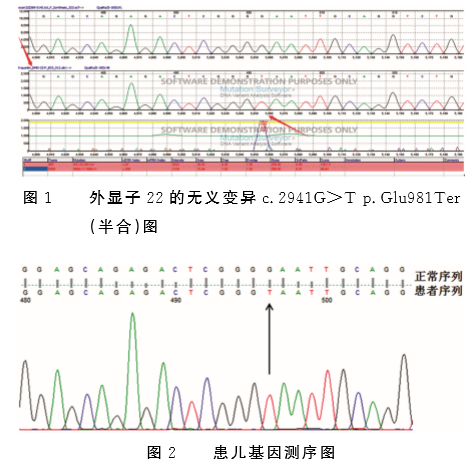
2 讨 论
本病例具有DMD的几个典型特征[ 1 3]:( 1)临床 特征:走路易疲劳,双侧腓肠肌假性肥大, Gowe r s征 阳性,下阶梯及快步行走呈典型鸭步;( 2)血清学检 测:心肌酶谱显著升高,ALT、AST 长期在200~ 700 U/L,CK 43 481 IU/L,CK-MB 1 114 IU/L, LDH 3 129 IU/L, α -HBDH 1 814 IU/L;( 3)肌电图 检查:肌电图提示右上肢三角肌、右下肢股四头肌静 息状态见异常自发电活动;( 4)基因检测:外显子22无义变异c. 2941G>T p. Gl u981Te r,可导致蛋白质 编码提前终止。基于以上几点,该患儿可明确诊断为 DMD。但该患儿确诊耗时近6个月时间,主要原因可 能有以下几方面原因:( 1)家庭重视不够:患儿平常身 体无明显不适,但走路易疲劳,仰卧需单手撑地才能 站立等症状早有出现,但患儿年龄尚小,被认为发育 迟,并未考虑存在疾病而被忽视未就医;( 2)临床医生 经验不足:该患儿因体检发现肝功能异常入院,一段 时间仅关注肝脏功能及相关检查较为局限,加之巨细 胞病毒阳性,而忽略其他如心肌酶谱、体格检查等; ( 3)患儿运动发育基本正常,会独立行走;( 4)无特殊 家族史,其同母哥哥体健。这些原因均导致了该患儿 疾病的延迟确诊。 DMD遗传方式为X连锁隐性遗传,发病率在各 个国家、地区和人种间无明显差异,每3 600~6 000 例出生 男 婴 中 有1例 发 病。中 国 的 发 病 率 约 为 1/3 853,估算全国患者约70 000人[ 4]。该病常于2~ 5岁起病,常于20岁左右死于心力衰竭或呼吸功能不 全[ 5]。该病在诊疗过程中重点要与其他类型肌营养 不良、脊肌萎缩症、炎性肌病和代谢性肌病进行鉴别 诊断,但通过临床表现、血肌酶、肌肉活检和基因检测 能够进行鉴别[ 4]。 DMD基因主要有3种突变类型[ 6 8]:( 1)大片段 缺失型:最为常见,突变发生频率约占所有突变的 60%;( 2)大片段重复型:较少见,约占所有突变的 10%;( 3)微小突变:包括单个或数个核苷酸置换、缺 失或插入等,约占所有突变的 30%。本病例应用 MLPA技术检测大片段缺失和大片段重复,结合二代 测序技术对DMD基因外显子及周围内含子进行微小 突变分析。能够明确病因,但目前尚无有效的治疗方 法。对其鉴别诊断,对患者的康复治疗有一定的指导 作用,同时,能够提供一定的遗传咨询与指导。
通过对本例DMD的确诊,提示临床在诊疗过程 中,对不明原因的肝功能异常,且经抗病毒、护肝等治 疗无效者,要加强体格检查及其他辅助检查,如血清 肌酶、肌电图及肌活检,必要时行基因检测,提高罕见 病的诊疗水平。
参考文献略。
原始出处:
杜 娟,刘星鹭,陈 诚,左江成等,Duchenne型肌营养不良1例诊断体会[J],国际检验医学杂志2 0 2 0年2月第4 1卷第3期
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言






#杜氏肌营养不良症#
133
#Chen#
69
#肌营养不良#
67
#Duchenne型肌营养不良#
68