ADC有效载荷的研究前沿
2022-12-01 小药说药 小药说药 发表于安徽省

抗体偶联药物(ADC)是由靶向特异性抗原的单克隆抗体与小分子细胞毒性药物通过连接子链接而成,兼具传统小分子化疗的强大杀伤效应及抗体药物的肿瘤靶向性。
前言
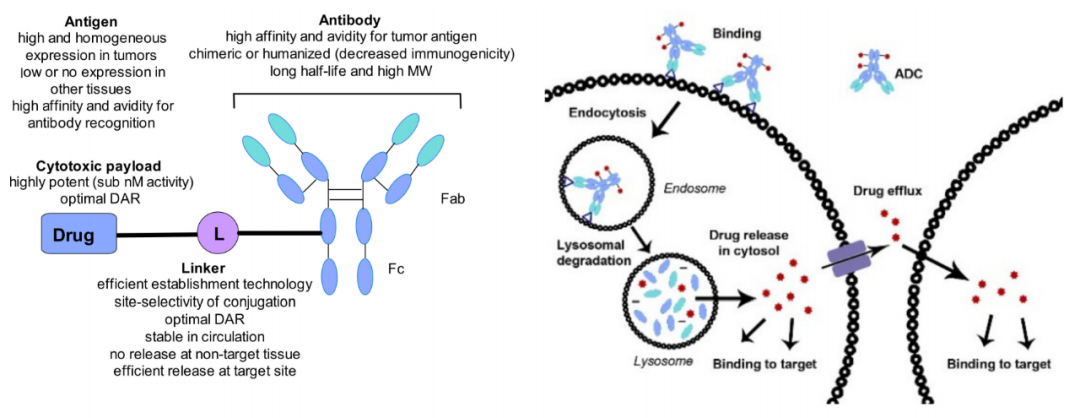
抗体偶联药物(ADC)是由靶向特异性抗原的单克隆抗体与小分子细胞毒性药物通过连接子链接而成,兼具传统小分子化疗的强大杀伤效应及抗体药物的肿瘤靶向性。ADC由三个主要部分组成:负责选择性识别癌细胞表面抗原的抗体,负责杀死癌细胞的药物有效载荷,以及连接抗体和有效载荷的连接子。
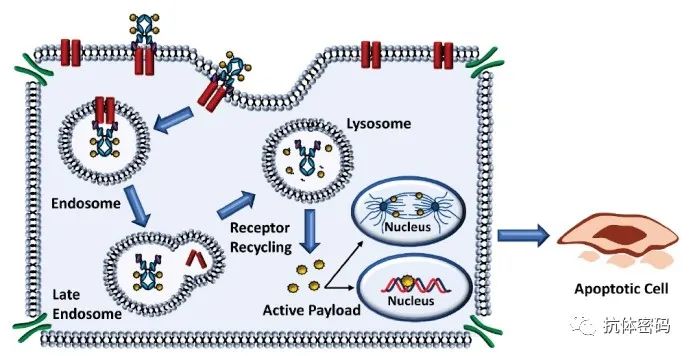
ADC对抗原的识别导致ADC通过内吞途径进入细胞内,通过溶酶体降解后,有效载荷以生物活性形式释放并发挥作用,导致癌细胞死亡。细胞内有效载荷的数量由每个细胞表面抗原的数量、每个ADC的药物有效载荷分子的数量(也称为药物抗体比率,DAR)以及抗原返回细胞表面所需的时间决定。有效载荷可能在癌细胞死亡和降解后逃逸,也可能从胞浆中透膜而出。这种释放的后果可能是有益的(也称为旁观者效应),也可能是有害的,导致全身毒性。
ADC中有几个变化的部分,成功显然没有通用的公式。因此,如何选择合适的抗体、在何处以及如何将连接子连接到抗体、每个抗体连接多少药物分子、如何连接连接子和药物有效载荷、最佳药物有效载荷是什么样的?这些问题需要我们深刻理解ADC各个组分的生物学和化学特性,才能获得满意的答案。
微管破坏药物
金盏花素
Auristatins是ADC中使用的重要有效载荷,最著名的家族成员MMAE存在于两种上市药物Adcetris和Polivy中。目前,超过10种以金盏花素(如MMAE)或一甲基金盏花素F(MMAF)为有效载荷的ADC正在进行临床试验。

上图描述了auristatine及其常用的连接位点。金盏花素的构效关系(SAR)已被广泛研究,主要集中在末端亚单位:P1(N-末端)和P5(C-末端),最常见的方法是在P1上引入氨基甲酸酯功能。
2015年,西雅图遗传学的研究人员将ADC有效载荷的范围扩大到包括叔胺,特别是N-二甲基auristatine,首次通过铵键将药物与单克隆抗体结合。所得ADCs在生理条件下稳定,体内外活性高,免疫特异性强。这些结果扩大了ADC可用于靶向给药的药物种类。
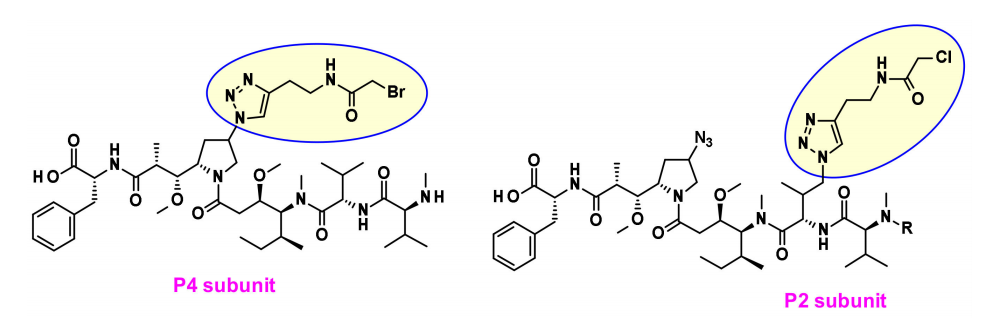
最近,Agensys公司通过调节中心亚基P2-P3-P4,将叠氮化物基团引入P2和P4亚基,在与蛋白酶可裂解的连接子偶联后,产生了在体外和体内效力提高的亲水性衍生物,这为连接子的连接提供了新途径。
一般来说,在同时含有胺和醇反应的auristatin中,首选的连接点是胺通过氨基甲酸酯键偶联。西雅图遗传学开发了一种新的策略,将含醇的有效载荷与亚甲基烷氧基氨基甲酸酯(MAC)偶联。为了稳定MAC键,碱性基团和吸电子基团都靠近氨基键,结果表明,该偶联物在生理条件下是稳定的,具有很高的效价,并且在体内外都具有免疫特异性。
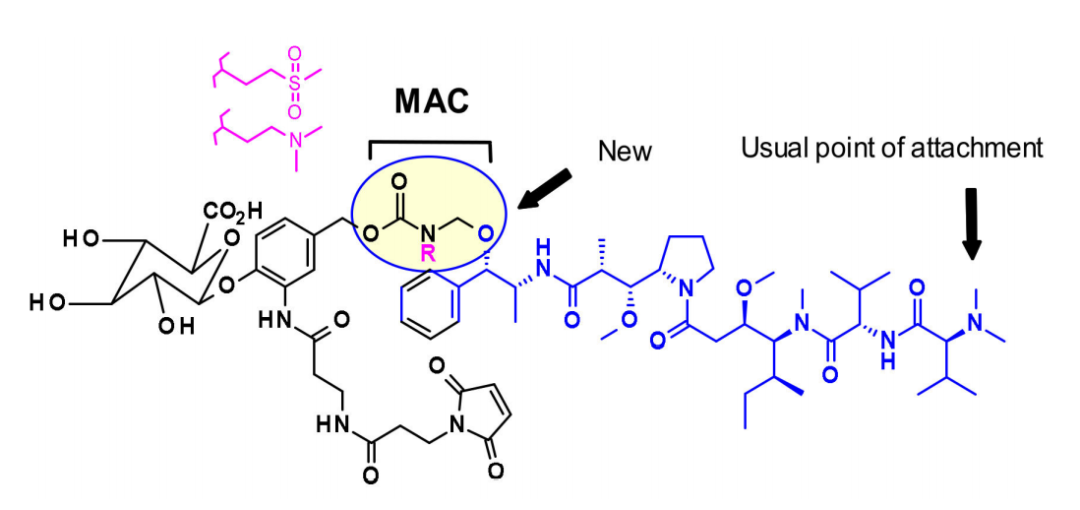
此外,乌普萨拉大学的研究人员还开发了AZASTATIN,作为一类新的强有力的auristatin衍生物,包含一个中心胺侧链的抗体结合位点。他们的研究结果证实,这些auristatin衍生物是一类新的细胞毒性有效载荷,适合ADC开发。

美登素衍生物(DM2,DM4)
美坦辛是一种非常有效的微管组装抑制剂,可诱导细胞的有丝分裂停止。但是这种结构很难共轭,因为它没有反应性官能团,为了克服这个问题,一系列含有SMe基团的非常有效的衍生物被创造出来。这一类分子的第一个例子是DM1和DM4,它们带有甲硫丙酰基而不是天然N-乙酰基。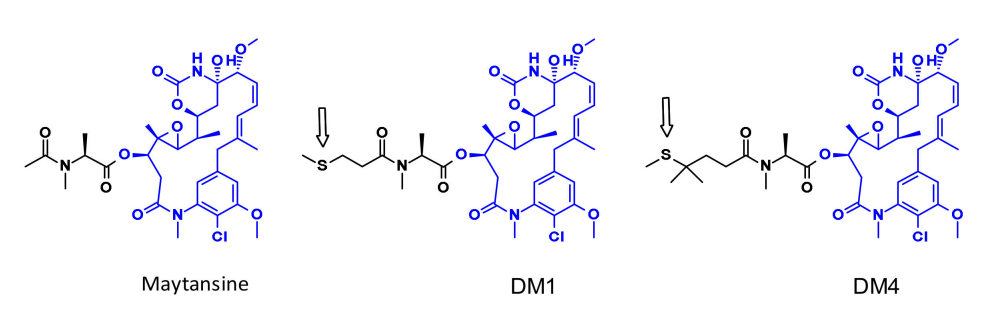
从有效载荷DM1和DM4中,通过使用二硫键与连接子偶联。稳定的二硫键连接子在血液循环中表现出良好的稳定性,同时在细胞内保持有效的分裂。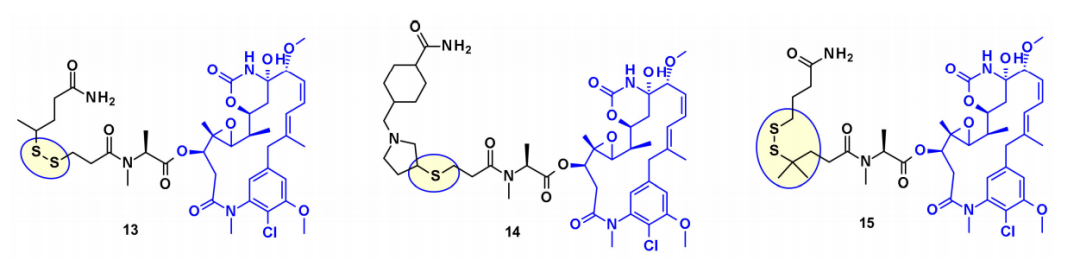
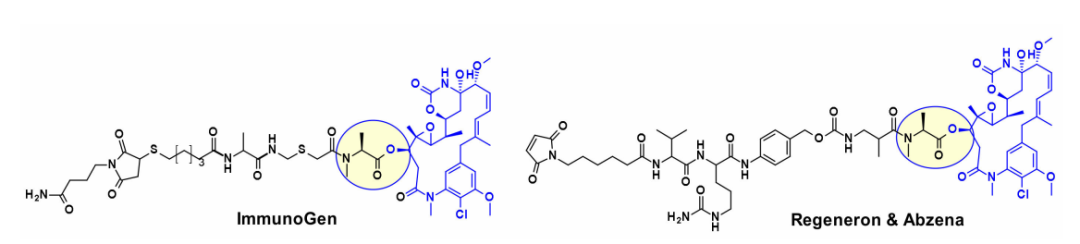
此外,几种基于maytansine的ADC利用相同的二级羟基作为附着点,并在大多数情况下携带转谷氨酰胺酶生物结合的连接子。例如,一种基于Daratumumab的ADC被证明能向CD38过度表达的癌细胞特异性地传递DM4。最近ImmunoGen开发了一种新型的ADC,它包含一种含硫的maytansinoid,通过一种高度稳定的三肽连接体连接到抗体上,附着点与上述羟基相同。与先前的美登素ADC相比,增加连接物中亚甲基单元的数量增加了旁观者杀伤活性,并提高了小鼠体内的疗效。在类似的方法中,保持核心大环不变,Regeneron和Abzena的研究人员研究了N-甲基丙氨酸氮取代的影响,也改变了大环上侧链的长度,以及通过伯胺和仲胺连接的连接子。
 微管溶素
微管溶素
Tubulysins是微管聚合的有效抑制剂,可导致分裂细胞的细胞骨架迅速解体,并导致细胞凋亡。它们是一个天然存在的四肽家族,含有Mep、Ile、Tuv和Tut,R3=OH或Tup,R3=H。
利用Tubulysins作为ADC有效载荷,其广泛的附着点已被充分开发。这种结构中一个明显的附着点是Tut或Tup模块的羧酸,如Endocyte的EC1428,其中羧酸通过酰肼部分连接到连接子。Oncomatryx公司也采用了同样的方法,以同样的方式安装了可切割的PABAValCit马来酰亚胺连接子。
阿斯利康、百时美施贵宝和辉瑞使用的另一种方法依赖于Tup或Tut中苯环的衍生化。
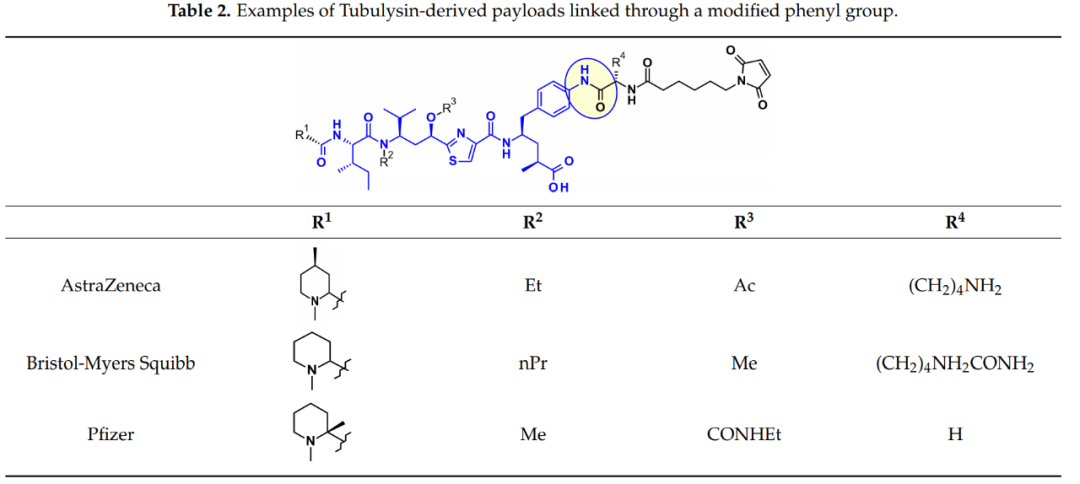
连接子与Mep基团的连接也得到了广泛的研究。Ingenica研究人员报告说,去甲基Mep类似物保留了强大的细胞毒性活性,可被视为有价值的有效载荷,允许在仲胺上引入不可切割的马来酰亚胺己基连接子。
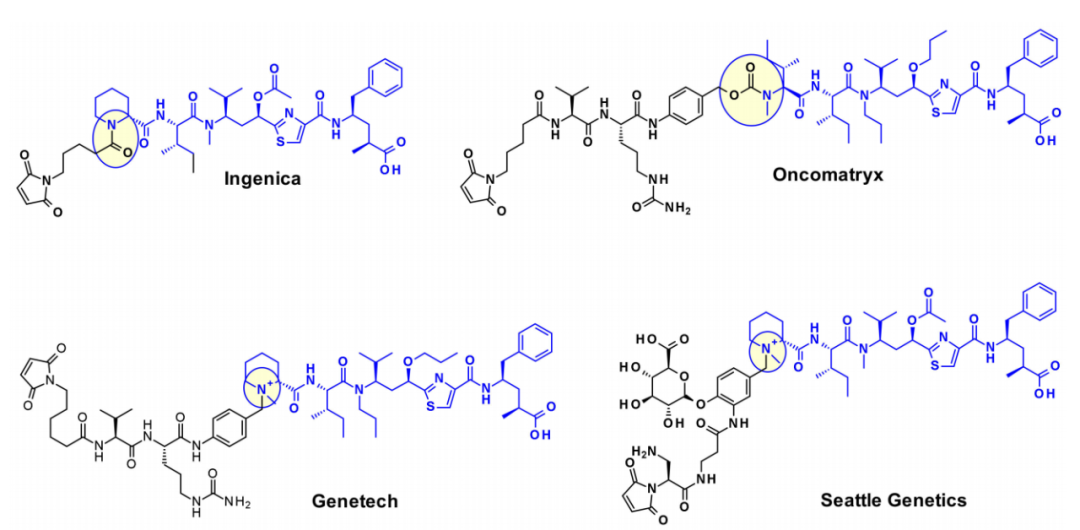
Oncomatryx的研究表明,当Mep被另一个拥有仲胺的基序取代时,通过氨基甲酸酯键引入可切割的连接体是产生ADC的有效途径。特别有趣的是,基因泰克关于通过季铵基团将连接子连接到含叔胺的有效载荷上。在有效载荷上引入mc-Val-Cit-PABA连接子导致更多亲水性结合物并改善了血液中的稳定性。西雅图遗传学也采用了同样的方法,通过过度表达葡萄糖醛酸,葡萄糖醛酸连接子也可以改善亲水性和选择性的细胞内切割通过癌细胞中过表达的β-葡萄糖醛酸酶。
隐粘菌素
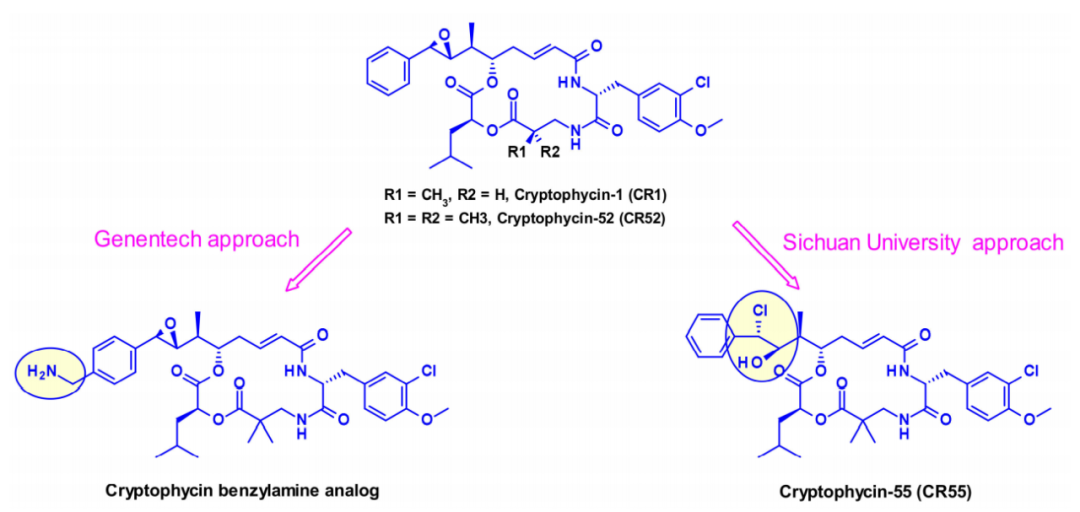
隐粘菌素(Cryptomycins,CR)是一个具有抗肿瘤活性的六元大环二肽家族。已有的临床试验的结果表明,在达到治疗效果所需的剂量下,其毒性水平是不可接受的。
几个小组尝试将CR用于ADC,但是由于在CR中缺少偶联位点,目前,有两种不同的方法通过引入其它基团,使得能够连接ADC的连接子。一种是由基因泰克的研究人员将苯转化为苄胺以产生有效的有效载荷,这种有效载荷适合通过氨基甲酸酯键连接。在第二种方法中,四川大学的研究人员利用了隐霉素-52的前药形式(CR55),它可以在生理条件下重新环化为CR52。
抗有丝分裂EG5抑制剂
纺锤体驱动蛋白(KSP,也称为Eg5或KIF11)是一种ATP依赖性运动蛋白,参与细胞周期中心体的分离。因此,用KSP抑制剂(KSPis)阻断有丝分裂中的这一重要事件可产生抗肿瘤效力。
拜耳发现了一个新的吡咯亚类的KSPis,他们研究了该分子不同位置与保持对KSP强亲和力的连接子的连接兼容性。
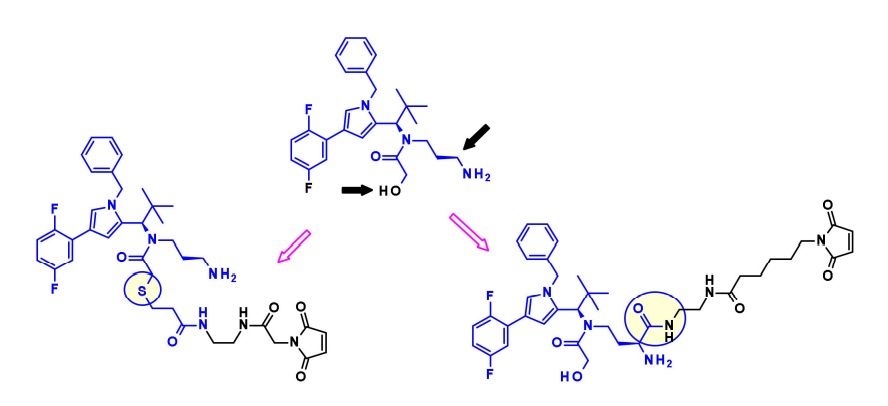
同样,诺华的研究人员使用含咪唑的KSP抑制剂作为Eg5 ADC。利用伯醇或仲酰胺部分,他们安装了带有马来酰亚胺端基的不可裂解连接子。当与靶向HER2和c-KIT的抗体偶联时,得到的ADC显示出优于Kadcyla的体内疗效。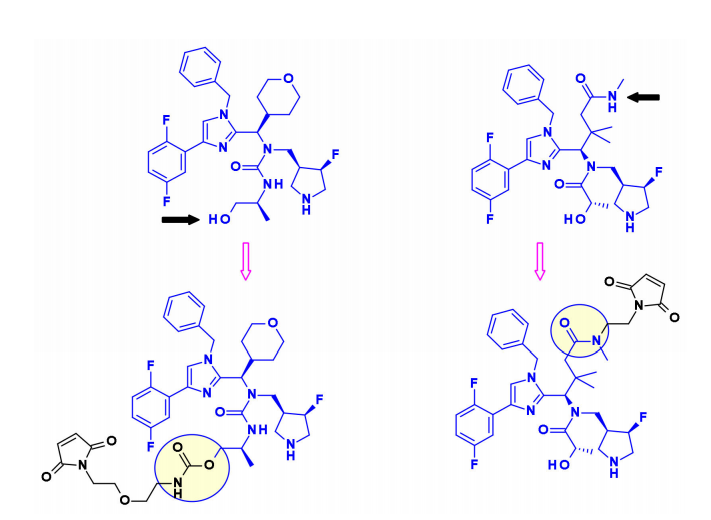 DNA损伤药物
DNA损伤药物
吡咯苯并氮卓类和吲哚氯苯并氮卓类
吡咯并[2,1-c][1,4]苯二氮杂卓(PBD)是一类具有抗肿瘤活性的天然产物。它们的作用方式是在DNA的小凹槽中进行选择性烷基化,其中鸟嘌呤的N2与PBD上的亲电N10/C11亚胺形成共价键。
西雅图遗传学使用SGD1882的苯胺作为附着点,模仿可切割连接子中常用的PAB单元,释放自由的PBD有效载荷。StemCentrx与Spirogen合作,利用PBD的N-10位置连接一个氨基甲酸酯的连接子。同样的氨基甲酸酯键也被Immunogen用于结构相似的吲哚氯苯偶氮卓二聚体(IBD)有效载荷。他们还报道了同一类IBD的不同方法,其中一个取代的苯环被用作两个IBD单体的C8/C8'位置之间的连接物。
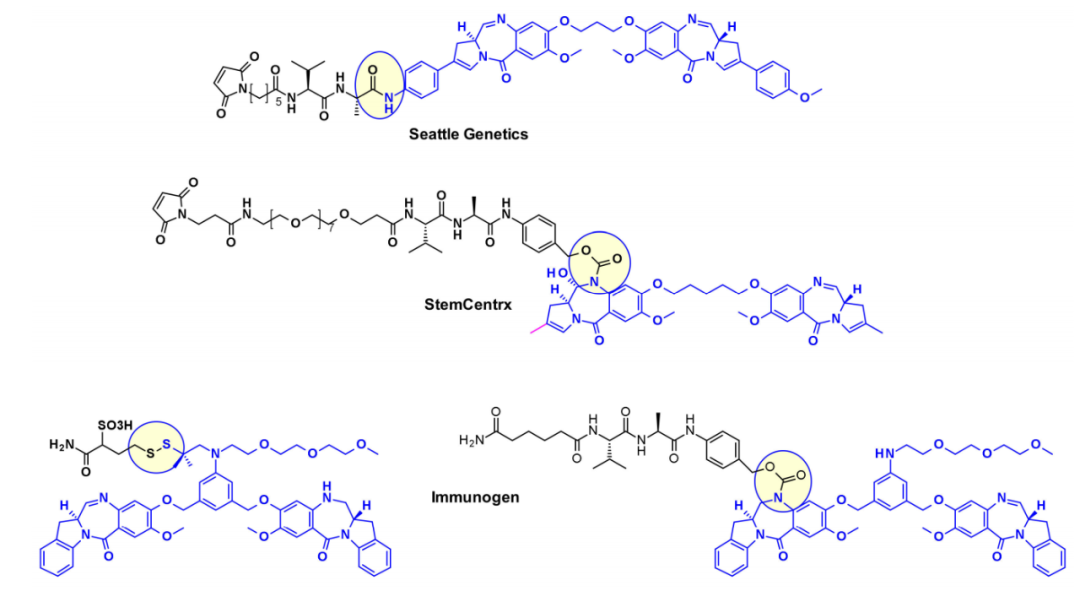
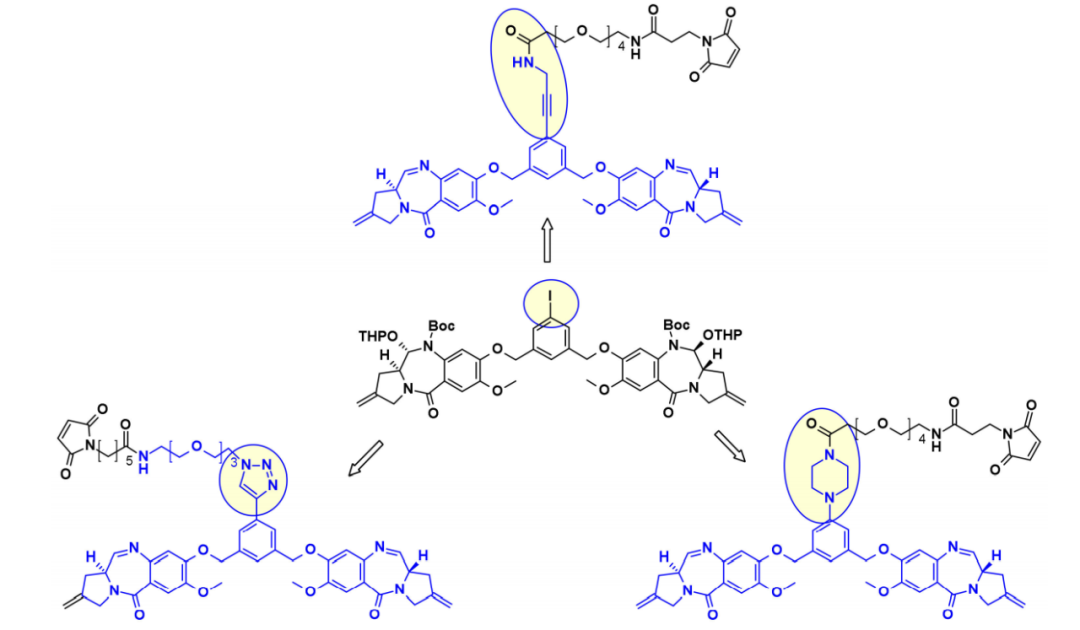
以类似的方法,Spirogen和Genentech设计了一种碘苯连接的PBD,允许在过渡金属催化反应中引入不同的连接子。通过使用Sonogashira偶联、Buchwald–Hartwig偶联或叠氮化物-炔烃点击反应,分别获得炔烃、哌嗪或三唑连接的连接子有效载荷共轭物。
 杜卡霉素
杜卡霉素
杜卡霉素是一种强大的细胞毒性物质,通过其高活性的环丙烷环与DNA的小凹槽结合,并在N3位置烷基化腺嘌呤。非环化的,卤甲基形式的杜卡霉素细胞毒性活性显著降低。由于分子中苯酚基团可作为内消旋体激活剂,从而形成亲电环丙烷,因此杜卡霉素ADC开发中的连接策略集中于酚官能团的连接子连接。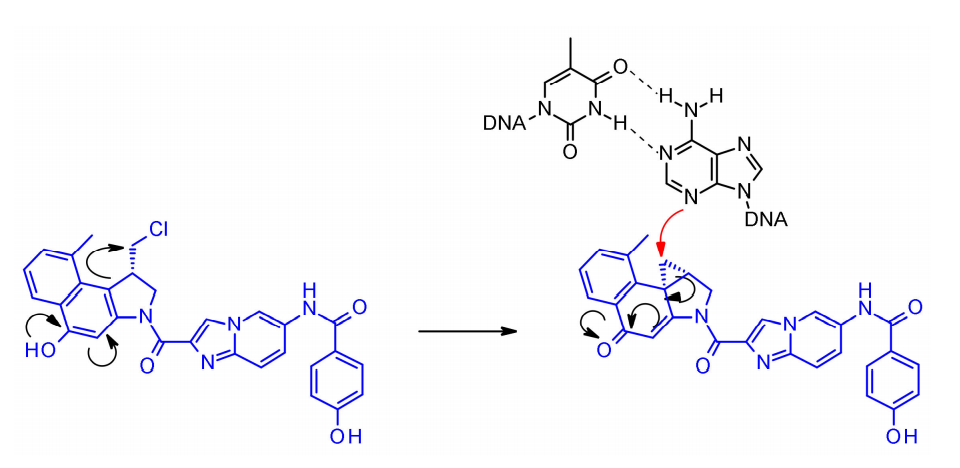
在Synthon开发的SYD985中,苯酚基团是通过双氨基甲酸酯连接连接子与Mc-val-cit-PABC有效载荷的位置。组织蛋白酶B裂解后,游离苯酚促进分子内重排成亲电环丙基形式。Medarex采用了一种不同的方法,通过分子非烷基化部分的芳香胺连接连接子,并用N-甲基哌嗪氨基甲酸酯部分掩蔽苯酚前药。在体内,苯酚将被释放,随后活性环丙烷将在羧酸酯酶的作用下形成。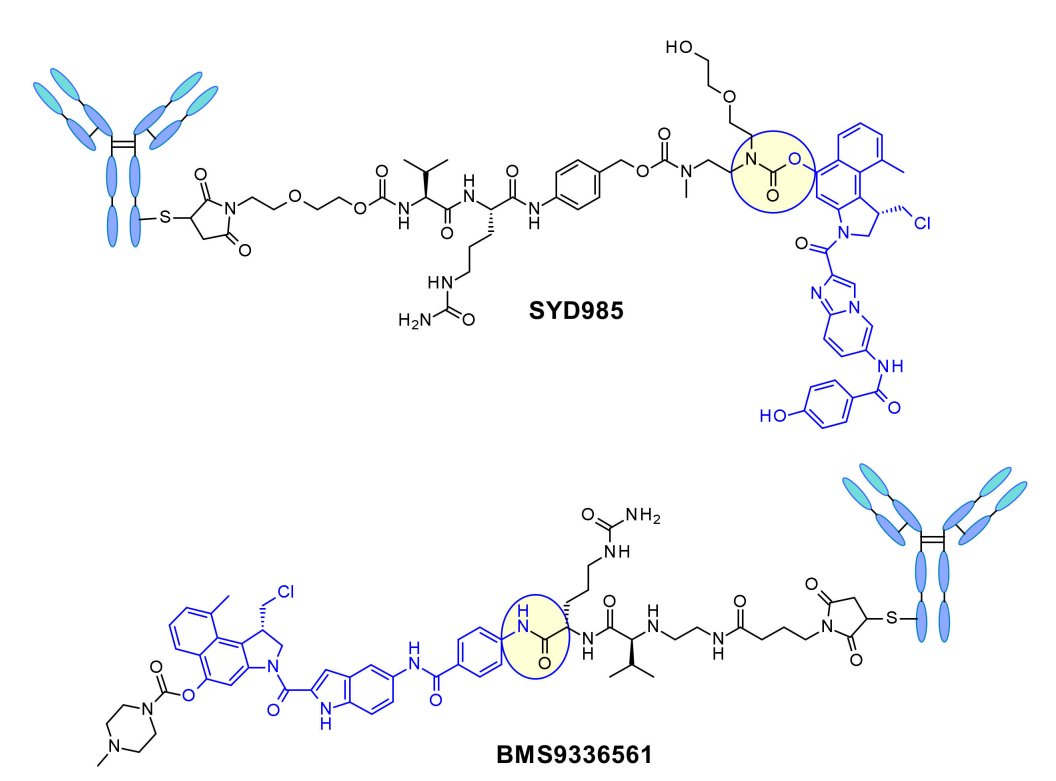 喜树碱
喜树碱
喜树碱(CPT)及其衍生物是拓扑异构酶I抑制剂的经典例子。它们稳定了拓扑异构酶诱导的DNA单链断裂,当三元DNA-TOP1-抑制剂复合物遇到复制叉时,DNA发生双链断裂。天然喜树碱是一种五环结构,其极低的溶解性阻止了其作为癌症治疗药物的广泛应用。其水溶性前药伊立替康获得了转移性结直肠癌的上市许可。SN-38是伊立新坦的活性代谢物,通过人体肝脏羧酸酯酶的作用在体内生成,其可通过打开内酯环而失活。
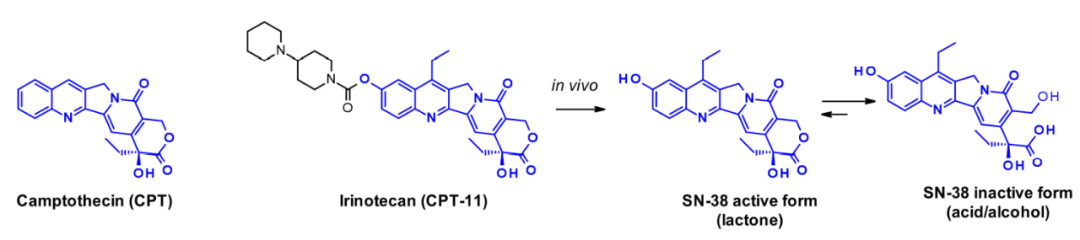
Immunomedics建立了两种不同的策略,通过其羟基部分结合SN-38。在一个例子中,连接子通过反应性更强的C-10苯酚基团连接,从而产生稳定的氨基甲酸酯键,而在另一个例子中,通过C-20羟基,同时稳定内酯形式,而C-20羟基对体内效力至关重要。

另一种适合ADC的非常有效的药物是依沙替康(DDX-8951f)。这种喜树碱类似物在其环己烷环上具有胺取代基,桥接7和9位。依沙替康的氨基有助于其水溶性,而环己烷环赋予的刚性被认为有利于活性内酯形式与非活性水解羟基酸的平衡。氨基羟基乙酰化生成DXd(1),而4-氨基丁酰化生成DXd(2),这两种化合物都保留了依沙替康的生物活性。

悬垂的羟基和氨基是明显的附着点,可使用酶可切割的Gly-Gly-Phe-Gly四肽连接子连接有效载荷。与抗HER2抗体偶联产生的ADC在临床环境中显示出对抗HER2表达癌症的巨大潜力。

DXd的环己胺环虽然被认为稳定了生物活性内酯形式,但它携带了一个手性中心,使合成工作和SAR研究复杂化。为了克服这一困难,Immunogen的研究人员研究了一组新的喜树碱类似物,这些类似物能够与单抗偶联。并且在这里,这个环被打开,额外的手性中心被消除。从一种常见的中间产物开始,研究人员尝试了三种类型的结构,并随后使用不同的聚苯胺连接子连接有效载荷。当与抗人表皮生长因子受体(HuEGFR)的抗体偶联时,产生的ADC对EGFR阳性的HSC-2肿瘤异种移植模型有效。 卡奇霉素
卡奇霉素
卡奇霉素是一类被广泛研究的烯二炔类抗生素,其结构和作用机制特别有趣和复杂,使其成为ADC有效载荷领域的一类抗生素。在ADC中连接calicheamicin的策略以市场上的adc Mylotarg为例,还有Besponsa。
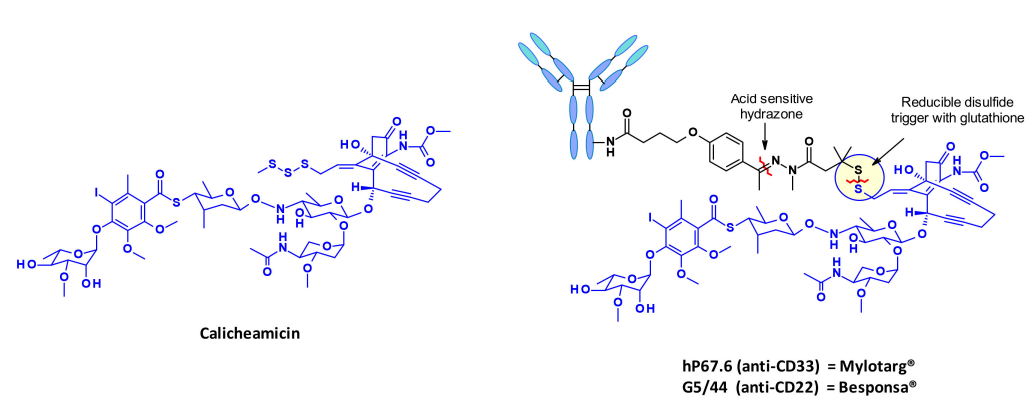
有效载荷的释放分两步进行:在酸性细胞内环境中对腙进行敏感的裂解,然后通过细胞内谷胱甘肽还原二硫键。释放的硫醇发生分子内1,4-加成的烯酮触发伯格曼环化反应,产生一个二自由基。这种活性中间体能够从脱氧核糖骨架中提取氢原子,产生双链DNA断裂,进而导致细胞死亡。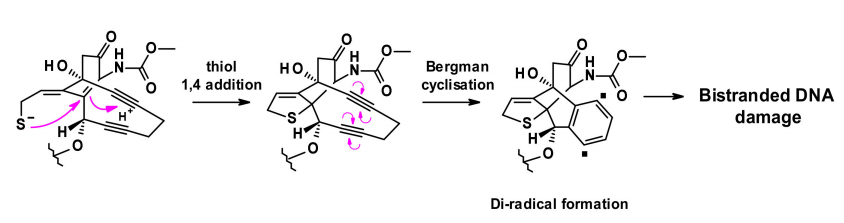
最近,从不列颠哥伦比亚地衣中发现的链霉菌中分离出一种新的烯二炔类天然产物,称为uncialamycin。这种结构通过全合成得到证实,从那时起,一些高效的合成类似物被制备成ADC的潜在有效载荷。
BMS的研究人员表明,由于在各种肽偶联条件下反应活性较低,因此,Uncialamycin的仲胺不是一个合适的连接点。研究人员合成了一种类似物,其中一个氨基直接引入到芳香环上,但这种苯胺的反应性也太弱,不能作为连接子引入的基团。另一方面,使用氨基乙基延伸物安装脂肪族胺,为连接子提供了合适的连接点。

从后一个有效载荷中,他们使用蛋白酶可切割的二肽和不可切割的连接子制备前体。CD70-ADC具有可切割连接体,对肾癌细胞株具有高度特异性的细胞毒活性,而相应的不可切割ADC在同一细胞株上不具有活性。

最近,为了继续这项工作,BMS的研究人员在设计的、高效的、化学稳定的uncialamycin类似物中使用苯酚基作为附着点。使用新开发的苯酚烷基化,在有效载荷的苯酚基团上添加了一个经典的可裂解连接子。将产生的有效载荷与抗体偶联,其在体外和体内均显示抗原特异性抗肿瘤活性。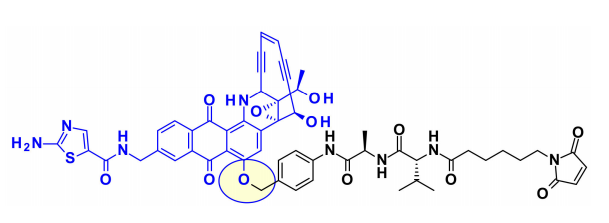 创新药物
创新药物
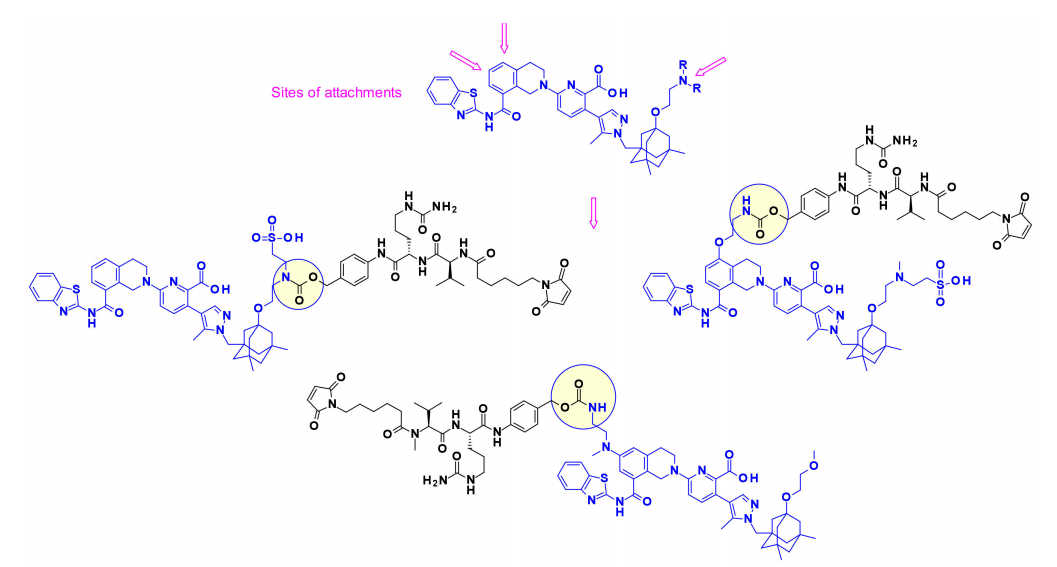
凋亡诱导剂(Bcl-xL抑制剂)
抗凋亡Bcl-2家族成员(包括Bcl-xL)的过度表达是癌细胞获得凋亡抵抗的机制之一。能够阻断Bcl-xL上BH3结合域的药物可以触发癌细胞凋亡。2017年,AbbVie首次以ADC的形式展示了BcL-xL抑制剂的有效载荷,其靶向表达EGFR的特定细胞或组织。有趣的是,研究人员在有效载荷上使用了三个不同的连接点来连接可切割的连接子。氨基烷基延伸的核心修饰用于在需要时建立合适的连接位点。
泰兰司他丁及其类似物
靶向剪接体是一种参与mRNA加工的大型核糖核蛋白复合物,为靶向癌症治疗提供了一种有希望的治疗选择。有几种天然产物能够通过与不同的剪接体亚单位结合来抑制RNA剪接。最具代表性的是thailanstatin A,它可以与剪接体的SF3b亚单位结合,从而阻止RNA剪接。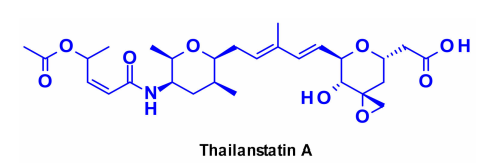
Thailanstatin A缺乏一个适合连接连接子的基团。为了解决这个问题,将羧酸与乙二胺偶联以引入含有胺的间隔基,该间隔基通常用于连接子的安装。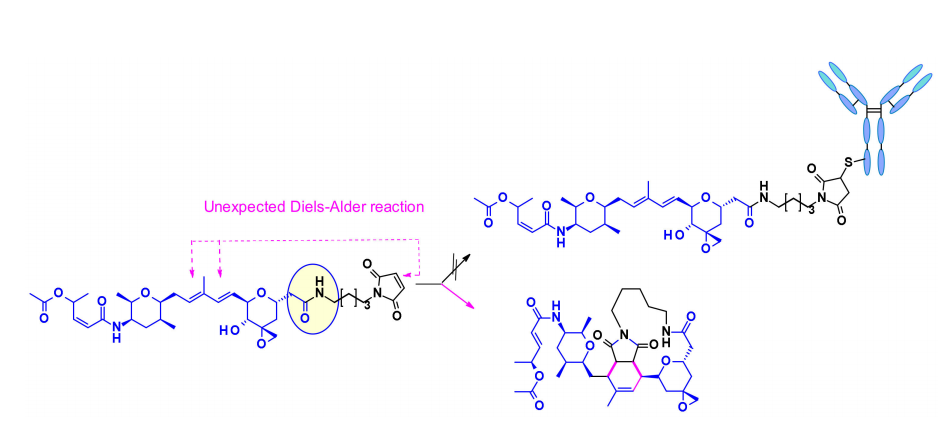
将这种天然产物用于ADC的另一个困难是存在多种反应性功能。例如,中心核中的二烯可以通过Diels–Alder反应与用于生物结合的马来酰亚胺部分反应。这个问题是通过使用另一个共轭部分,卤代乙酰胺来解决的。结合这两种修饰并包含可切割连接子的ADC首次在专利文献中报道,并且声称它们在几个表达HER2的细胞系中具有适度的活性。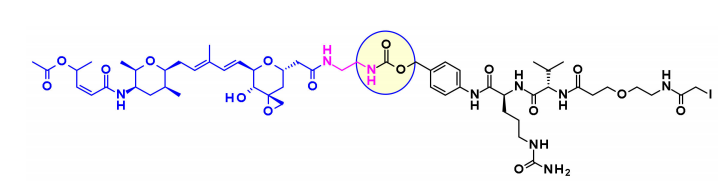
最近,辉瑞报告说,羧酸直接与抗体的有效表面赖氨酸(无连接子偶联)结合导致迄今为止最有效的Thailanstatin ADC。这些赖氨酸偶联物的活性与药物负载有关,而其他有效载荷类通常没有观察到这种特性。ADCs在胃癌异种移植模型中显示出良好的作用。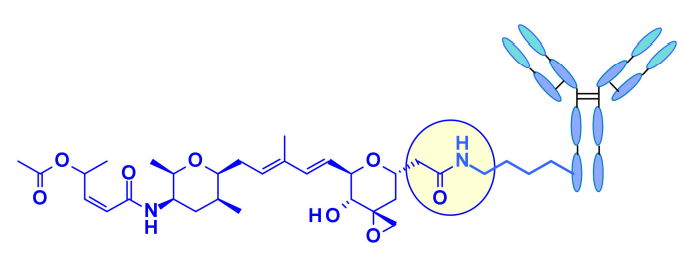 鹅膏毒素
鹅膏毒素
在ADC技术领域,使用类似amatoxins的转录抑制剂是一种相对较新的方法。九种天然存在的amatoxin衍生物具有相同的骨架结构,一个由八个L-构型氨基酸组成的大环,通过亚砜部分连接在色氨酸和半胱氨酸残基之间。amatoxins的三个侧链是羟基化的,OH基团具有良好的水溶性并与目标分子结合。两种肽,α-鹅膏糖蛋白和β-鹅膏毒素,占所有毒素的90%。

在amatoxins上共使用过三个附着点产生ADC。第一次尝试是将β-鹅膏毒素的羧基偶联到IgG上赖氨酸的氨基,这种连接具有良好的血浆稳定性和高细胞毒性,但这种生物偶联的产率很低。二氢异亮氨酸的羟基也被认为是一个连接点,引入谷胱甘肽作为连接子,然后通过赖氨酸结合,可获得体外细胞毒性和体内抗肿瘤活性优异的ADC,但不幸的是,由于血清羧酸酯酶裂解连接子,导致其循环稳定性差。第三种方法,附着于色氨酸的6-羟基代表了目前的标准程序,苯酚与各种连接子的醚化导致了高度稳定和有效的ADC。由于鹅膏毒素其它氨基酸要么不具化学活性,要么是与RNA聚合酶II结合的关键,因此不能将其它的鹅膏毒素氨基酸(即羟脯氨酸、甘氨酸、异亮氨酸和半胱氨酸)用于偶联。
基于amanitin的代表性的ADC为HDP-101。有效载荷本身是一种合成的金刚烷醇衍生物,优化了稳定性。与天然天麻素相比,两个差异是色氨酸中没有6′-OH,硫醚链取代亚砜。通过天冬氨酸侧链上酰胺的形成,引入组织蛋白酶B-裂解连接子。
最近,Park及其合作者为含苯酚的有效载荷设计了一种新的连接基序(OHPAS)。它是一种二芳基硫酸盐,一个芳基部分来自有效载荷,另一个来自连接基序的潜在苯酚基团。将该技术应用于曲妥珠单抗ADC中的a-鹅膏毒素,在体外和体内表现出强大的细胞毒性。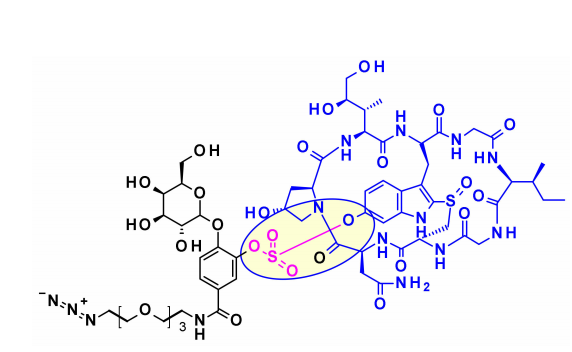
烟酰胺磷酸核糖转移酶
烟酰胺磷酸核糖基转移酶(NAMPT)是一种负责将烟酰胺转化为烟酰胺单核苷酸的酶,其抑制剂在各种临床前和临床研究中显示出有效性,但其临床应用受到靶向毒性和剂量限制性毒性的限制,如血小板减少和胃肠道不良反应。
诺华公司的研究人员在NAMPT抑制剂有效载荷中的苯环对位中引入哌嗪部分,确定了合适的连接子附着点。这种分子在c-Kit和HER2表达细胞系上表现出纳摩尔水平的效力,并且耐受性良好,在体内表现出靶向依赖性。
 卡马霉素
卡马霉素
从库拉索菌中分离得到两种新的蛋白酶抑制剂,卡马霉素A和卡马霉素B。两者都具有亮氨酸衍生的α,β-环氧酮弹头直接连接到甲硫氨酸亚砜或甲硫氨酸砜。他们被发现能抑制酿酒酵母20S蛋白酶β5亚单位活性(糜蛋白酶样活性)。此外,它们对肺癌和结肠癌细胞株具有很强的细胞毒性。
然而,由于它们的高效价,它们的选择性较差,经常表现出毒副作用。因此,剧毒的卡马霉素衍生物适合作为ADC的弹头,可以保持所需的效力,并获得更好的耐受性。所设计的类似物均在P2位置并入砜甲硫氨酸衍生物(如卡马霉素B),而不是卡马霉素A中的亚砜甲硫氨酸,因为这消除了亚砜基立体异构体混合物产生的结构复杂性。
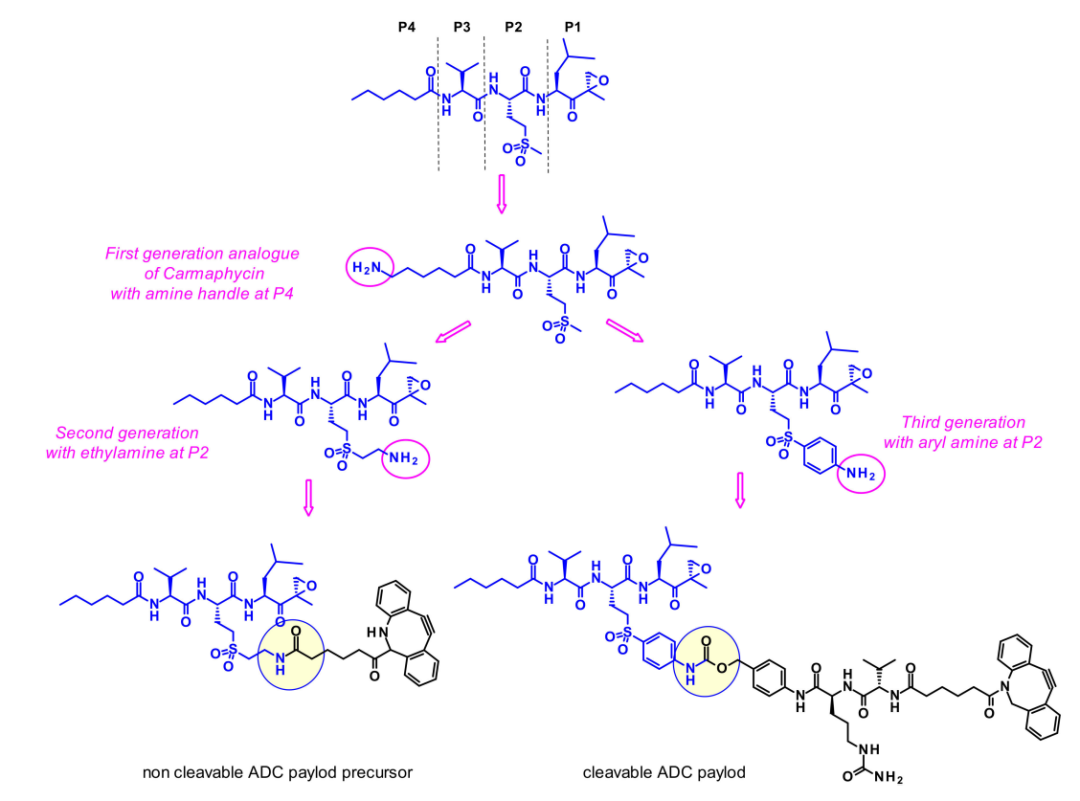
第一代类似物在P4末端含有胺基,不幸的是,这个附着点并不合适,因为有效载荷显示细胞毒性活性降低。第二代卡马霉素类似物在P2侧链上含有一个胺基,当短乙基氨基链延伸磺酰基时,效果最好。在第三代类似物中,芳基连接砜和胺,因此降低了它的碱性。第二代和第三代有效载荷都显示出强大的体外活性,并用可切割或不可切割的连接子连接。不幸的是,没有一种ADC对受试癌细胞系表现出比曲妥珠单抗更高的细胞杀伤能力。
本网站所有内容来源注明为“梅斯医学”或“MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明来源为“梅斯医学”。其它来源的文章系转载文章,或“梅斯号”自媒体发布的文章,仅系出于传递更多信息之目的,本站仅负责审核内容合规,其内容不代表本站立场,本站不负责内容的准确性和版权。如果存在侵权、或不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言